{"title":"Lifelong Machine Learning Potentials for Chemical Reaction Network Explorations","authors":"Marco Eckhoff*, and , Markus Reiher*, ","doi":"10.1021/acs.jctc.5c01127","DOIUrl":null,"url":null,"abstract":"<p >Recent developments in computational chemistry facilitate the automated quantum chemical exploration of chemical reaction networks for the in-silico prediction of synthesis pathways, yield, and selectivity. However, the underlying quantum chemical energy calculations require vast computational resources, limiting these explorations severely in practice. Machine learning potentials (MLPs) offer a solution to increase computational efficiency, while retaining the accuracy of reliable first-principles data used for their training. Unfortunately, MLPs will be limited in their generalization ability within chemical (reaction) space, if the underlying training data are not representative for a given application. Within the framework of automated reaction network exploration, where new reactants or reagents composed of any elements from the periodic table can be introduced, this lack of generalizability will be the rule rather than the exception. Here, we therefore evaluate the benefits of the lifelong MLP concept in this context. Lifelong MLPs push their adaptability by efficient continual learning of additional data. We propose an improved learning algorithm for lifelong adaptive data selection yielding efficient integration of new data while previous expertise is preserved. In this way, we can reach chemical accuracy in reaction search trials.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9641–9656"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/acs.jctc.5c01127","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01127","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
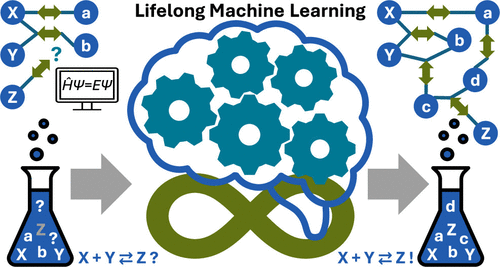
Abstract
Recent developments in computational chemistry facilitate the automated quantum chemical exploration of chemical reaction networks for the in-silico prediction of synthesis pathways, yield, and selectivity. However, the underlying quantum chemical energy calculations require vast computational resources, limiting these explorations severely in practice. Machine learning potentials (MLPs) offer a solution to increase computational efficiency, while retaining the accuracy of reliable first-principles data used for their training. Unfortunately, MLPs will be limited in their generalization ability within chemical (reaction) space, if the underlying training data are not representative for a given application. Within the framework of automated reaction network exploration, where new reactants or reagents composed of any elements from the periodic table can be introduced, this lack of generalizability will be the rule rather than the exception. Here, we therefore evaluate the benefits of the lifelong MLP concept in this context. Lifelong MLPs push their adaptability by efficient continual learning of additional data. We propose an improved learning algorithm for lifelong adaptive data selection yielding efficient integration of new data while previous expertise is preserved. In this way, we can reach chemical accuracy in reaction search trials.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


