Integrating a multitask graph neural network with DFT calculations for site-selectivity prediction of arenes and mechanistic knowledge generation
IF 20
0 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
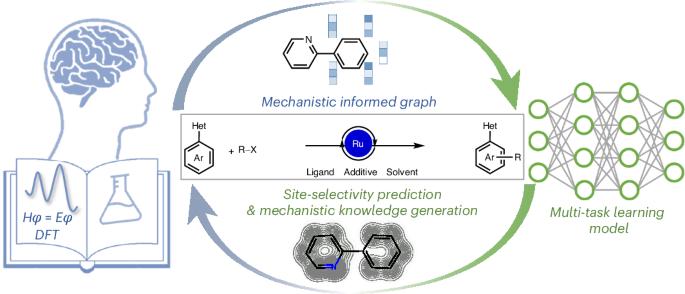
The accurate prediction of reaction performance based on empirical knowledge paves the way to efficient molecule design. Compared with the human-summarized reaction knowledge of a focal dataset, the machine-learned quantitative structure–performance relationship of larger-scale datasets is more effective at accessing the entire chemical space. Here we report a multitask learning workflow combined with a mechanism-informed graph neural network to predict site selectivity for ruthenium-catalysed C–H functionalization of arenes. The multitask architecture enables the acquisition of related knowledge from the simultaneous learning tasks. The embedded reaction graph bridges the gap between previous mechanistic studies and reaction representation. Along with this mechanistic embedding, the developed multitask model demonstrates excellent interpolative and extrapolative ability on the reported dataset composed of 256 reactions, achieving an average site-selectivity prediction accuracy of 0.934 with a standard deviation of 0.007. The prediction scope ranges from simple to fused arenes and was even extended to heterocyclic indole derivatives in the additional out of sample tests containing 14 unseen instances. Furthermore, interpretation of the model promotes the development of a para-selective mechanistic model verified by density functional theory calculations. A multitask graph neural network is developed with mechanism-informed reaction graphs for site-selectivity prediction of ruthenium-catalysed C‒H functionalization of arenes. The extrapolative prediction ability of the model is verified by experimental tests. Interpretation of the model deepens our understanding of the origins of the site selectivity.
将多任务图神经网络与DFT计算相结合,用于芳烃选择性预测和机理知识生成
基于经验知识的反应性能的准确预测为高效的分子设计铺平了道路。与人类对焦点数据集的总结反应知识相比,机器学习的大规模数据集的定量结构-性能关系在访问整个化学空间方面更有效。在这里,我们报告了一个多任务学习工作流程,结合机制信息图神经网络来预测钌催化芳烃C-H功能化的位点选择性。多任务架构使得从同步学习任务中获取相关知识成为可能。嵌入的反应图弥补了以前的机理研究和反应表征之间的差距。通过这种机制嵌入,所建立的多任务模型对256个反应组成的报告数据集表现出良好的内插和外推能力,平均选择性预测精度为0.934,标准差为0.007。预测范围从简单芳烃到融合芳烃,甚至在包含14个未见实例的额外样品外测试中扩展到杂环吲哚衍生物。此外,对该模型的解释促进了准选择机制模型的发展,并得到密度泛函理论计算的验证。提出了一种具有反应机理的多任务图神经网络,用于预测钌催化芳烃碳氢官能化反应的选择性。通过实验验证了该模型的外推预测能力。对该模型的解释加深了我们对位点选择性起源的理解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


