{"title":"Design of bimetallic single-atom catalysts and theoretical study of CO2 reduction reaction","authors":"Yang Liu, Zhao-Di Yang and Guiling Zhang","doi":"10.1039/D5NJ02459E","DOIUrl":null,"url":null,"abstract":"<p >The electrocatalytic conversion of CO<small><sub>2</sub></small> represents a promising strategy toward achieving carbon neutrality, where single-atom catalysts (SACs) with atomically dispersed metal centers demonstrate exceptional potential for the CO<small><sub>2</sub></small> reduction reaction (CO<small><sub>2</sub></small>RR). This work develops Co<small><sub><em>x</em></sub></small>Fe<small><sub>3−<em>x</em></sub></small>–O<small><sub>4</sub></small>–C<small><sub>4</sub></small>N SACs (<em>x</em> = 0–3) featuring tailored multi-metallic sites and systematically investigates their CO<small><sub>2</sub></small>RR pathways and competitive hydrogen evolution reaction (HER) pathway through density functional theory (DFT) calculations. Detailed analysis of key intermediates reveals that the Fe sites in Co<small><sub>1</sub></small>Fe<small><sub>2</sub></small>–O<small><sub>4</sub></small>–C<small><sub>4</sub></small>N SACs exhibit optimal performance for HCOOH production, achieving a remarkably low energy barrier of 0.51 eV at the rate-determining step. Comparative studies demonstrate that Fe incorporation effectively suppresses competitive HER at Co sites while simultaneously modulating the electronic structures and charge transfer ability, which optimizes intermediate adsorption configurations, particularly enhancing activity and selectivity toward HCOOH generation. The findings establish a fundamental framework for designing multi-metallic SACs with precisely engineered active sites and high selectivity for special high-value-added chemicals.</p>","PeriodicalId":95,"journal":{"name":"New Journal of Chemistry","volume":" 37","pages":" 16113-16121"},"PeriodicalIF":2.5000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/nj/d5nj02459e","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
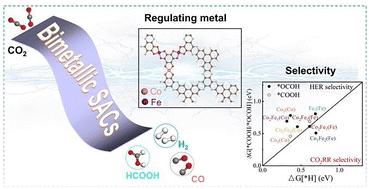
The electrocatalytic conversion of CO2 represents a promising strategy toward achieving carbon neutrality, where single-atom catalysts (SACs) with atomically dispersed metal centers demonstrate exceptional potential for the CO2 reduction reaction (CO2RR). This work develops CoxFe3−x–O4–C4N SACs (x = 0–3) featuring tailored multi-metallic sites and systematically investigates their CO2RR pathways and competitive hydrogen evolution reaction (HER) pathway through density functional theory (DFT) calculations. Detailed analysis of key intermediates reveals that the Fe sites in Co1Fe2–O4–C4N SACs exhibit optimal performance for HCOOH production, achieving a remarkably low energy barrier of 0.51 eV at the rate-determining step. Comparative studies demonstrate that Fe incorporation effectively suppresses competitive HER at Co sites while simultaneously modulating the electronic structures and charge transfer ability, which optimizes intermediate adsorption configurations, particularly enhancing activity and selectivity toward HCOOH generation. The findings establish a fundamental framework for designing multi-metallic SACs with precisely engineered active sites and high selectivity for special high-value-added chemicals.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


