Fibroblast-secreted ADAMTSL2 promotes cardiac repair after myocardial infarction by activating LRP6/β-catenin signaling
IF 3.7
2区 生物学
Q2 CELL BIOLOGY
引用次数: 0
Abstract
When coronary reperfusion is delayed in myocardial infarction (MI), persistent ischemic injury induces progressive loss of cardiomyocytes, ultimately resulting in pathological ventricular remodeling and heart failure. Secreted proteins may play a critical role in modulating cardiomyocyte death following MI, offering potential therapeutic targets. This study elucidated the biological function of fibroblast-secreted ADAMTSL2 in cardiomyocyte apoptosis post-MI. Elevated ADAMTSL2 levels were detected by ELISA in the serum of acute myocardial infarction patients and by immunoblotting in the infarcted myocardium of mice. Hypoxia treatment significantly upregulated ADAMTSL2 expression in neonatal rat cardiac fibroblasts (NRCFs), whereas no such hypoxic response was observed in neonatal rat cardiomyocytes (NRCMs). In vitro, overexpression of ADAMTSL2 in NRCFs attenuated oxygen-glucose deprivation (OGD)-induced apoptosis in NRCMs, whereas knockdown of ADAMTSL2 in NRCFs exacerbated cardiomyocyte apoptosis. In vivo, fibroblast-targeted overexpression of ADAMTSL2 by adenovirus 5 significantly reduced cardiomyocyte apoptosis and ameliorated adverse left ventricular remodeling post-MI. Conversely, ADAMTSL2 knockdown exacerbated infarct size and accelerated pathological remodeling. Mechanistically, ADAMTSL2 overexpression increased the expression of β-catenin in cardiomyocytes. Co-immunoprecipitation (Co-IP) assay showed that ADAMTSL2 could directly bind to LRP6 and promote its phosphorylation, leading to β-catenin stabilization and subsequent nuclear translocation. In summary, our study indicates that ADAMTSL2 protects against cardiomyocyte apoptosis and promotes cardiac repair by activating LRP6/β-catenin signaling.
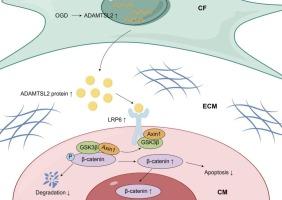
成纤维细胞分泌的ADAMTSL2通过激活LRP6/β-catenin信号通路促进心肌梗死后的心脏修复。
当心肌梗死(MI)的冠状动脉再灌注延迟时,持续的缺血性损伤会导致心肌细胞的进行性损失,最终导致病理性心室重构和心力衰竭。分泌蛋白可能在心肌梗死后心肌细胞死亡调节中起关键作用,提供潜在的治疗靶点。本研究阐明了成纤维细胞分泌的ADAMTSL2在心肌梗死后心肌细胞凋亡中的生物学功能。ELISA法检测急性心肌梗死患者血清中ADAMTSL2水平升高,免疫印迹法检测梗死小鼠心肌中ADAMTSL2水平升高。缺氧处理显著上调新生大鼠心脏成纤维细胞(nrcf)中ADAMTSL2的表达,而在新生大鼠心肌细胞(NRCMs)中没有观察到这种缺氧反应。在体外,nrcf中ADAMTSL2的过表达减轻了氧糖剥夺(OGD)诱导的NRCMs细胞凋亡,而nrcf中ADAMTSL2的下调则加重了心肌细胞凋亡。在体内,腺病毒5靶向成纤维细胞过表达ADAMTSL2可显著减少心肌细胞凋亡,改善心肌梗死后不良左心室重构。相反,ADAMTSL2敲低会加重梗死面积,加速病理性重构。机制上,ADAMTSL2过表达增加了心肌细胞中β-catenin的表达。共免疫沉淀(Co-IP)实验表明,ADAMTSL2可以直接结合LRP6并促进其磷酸化,导致β-catenin稳定和随后的核易位。综上所述,我们的研究表明,ADAMTSL2通过激活LRP6/β-catenin信号通路,保护心肌细胞凋亡,促进心脏修复。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Cellular signalling
生物-细胞生物学
CiteScore
8.40
自引率
0.00%
发文量
250
审稿时长
27 days
期刊介绍:
Cellular Signalling publishes original research describing fundamental and clinical findings on the mechanisms, actions and structural components of cellular signalling systems in vitro and in vivo.
Cellular Signalling aims at full length research papers defining signalling systems ranging from microorganisms to cells, tissues and higher organisms.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


