Toward standardized epitranscriptome analytics: an inter-laboratory comparison of mass spectrometric detection and quantification of modified ribonucleosides in human RNA.
Martin Hengesbach, Chi-Kong Chan, Tulsi Bhandari, Alan Bruzel, Michael S DeMott, Ganna Podoprygorina, Guangxin Sun, Ellen Tabeling, Vivian G Cheung, Peter C Dedon, Mark Helm, Patrick A Limbach
{"title":"Toward standardized epitranscriptome analytics: an inter-laboratory comparison of mass spectrometric detection and quantification of modified ribonucleosides in human RNA.","authors":"Martin Hengesbach, Chi-Kong Chan, Tulsi Bhandari, Alan Bruzel, Michael S DeMott, Ganna Podoprygorina, Guangxin Sun, Ellen Tabeling, Vivian G Cheung, Peter C Dedon, Mark Helm, Patrick A Limbach","doi":"10.1093/nar/gkaf895","DOIUrl":null,"url":null,"abstract":"<p><p>The human RNome comprises all forms of RNA and the 50 + chemical structures-the epitranscriptome-that modify them. Understanding the diverse functions of RNA modifications in regulating gene expression and cell phenotype requires technologies such as RNA sequencing-based modification mapping and mass spectrometry-based quantification of modified ribonucleosides. Liquid chromatography-coupled tandem quadrupole mass spectrometry (LC-MS/MS) is the gold standard for detecting and quantifying modified ribonucleosides with accuracy and precision. However, variations in RNA isolation, processing, and LC-MS/MS analysis have hindered reproducibility across laboratories, which is essential for accurate quantification of RNA modifications. As guidance toward harmonization, we report a multi-laboratory comparison of workflows for LC-MS/MS RNA modification analysis. We compared protocols for sample shipment, RNA hydrolysis, LC-MS/MS analysis, and data processing among three laboratories working with the same total RNA samples. We detected and quantified 17 modifications consistently across protocols and operators, with another 7 that were sensitive to experimental conditions, reagent contamination, and ribonucleoside instability, leading to poor precision among laboratories. Agreement among the three labs was strong, with coefficients of variation of 20% and 10% for relative and absolute quantification, respectively. These findings establish a robust and readily adoptable epitranscriptome analytical platform that enables reliable comparisons across laboratories.</p>","PeriodicalId":19471,"journal":{"name":"Nucleic Acids Research","volume":"53 17","pages":""},"PeriodicalIF":13.1000,"publicationDate":"2025-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12445667/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nucleic Acids Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/nar/gkaf895","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
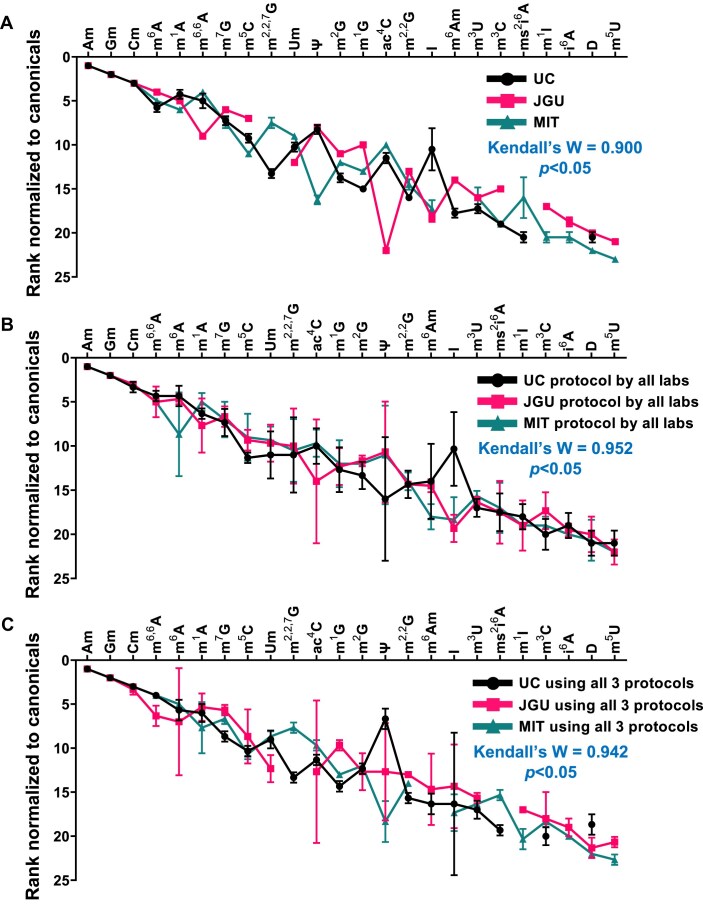
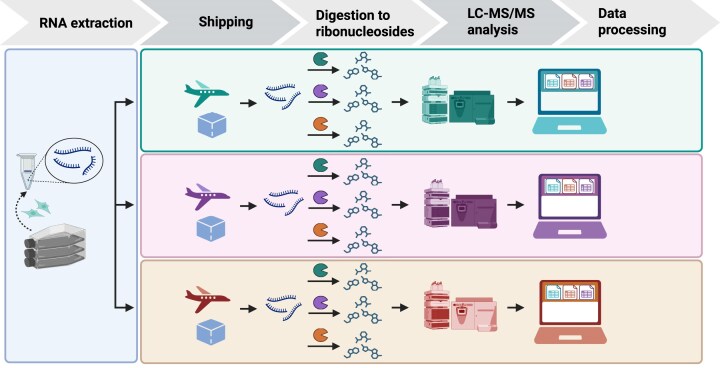
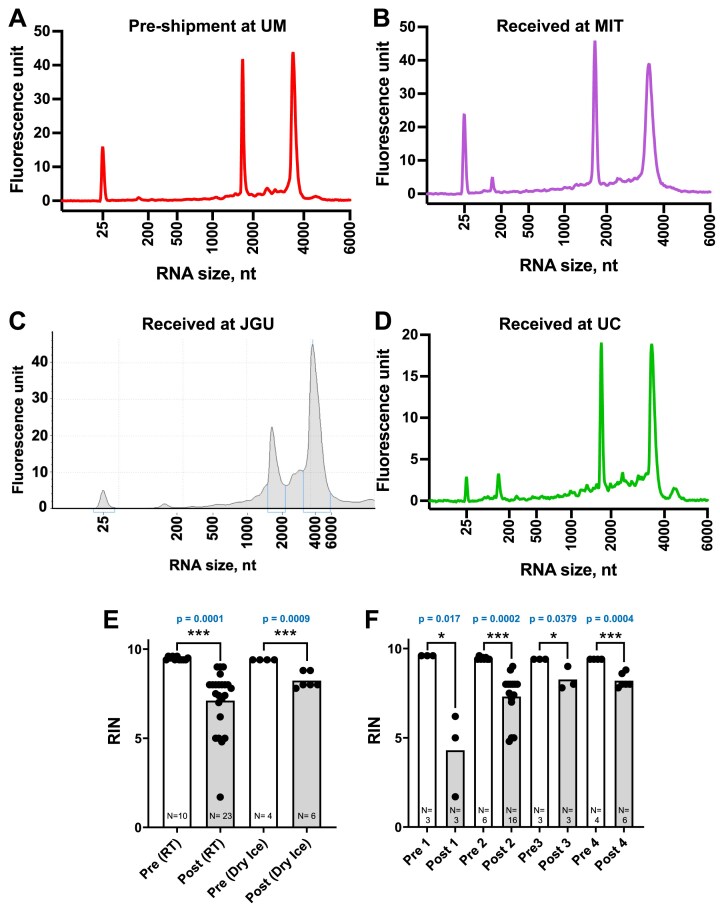
The human RNome comprises all forms of RNA and the 50 + chemical structures-the epitranscriptome-that modify them. Understanding the diverse functions of RNA modifications in regulating gene expression and cell phenotype requires technologies such as RNA sequencing-based modification mapping and mass spectrometry-based quantification of modified ribonucleosides. Liquid chromatography-coupled tandem quadrupole mass spectrometry (LC-MS/MS) is the gold standard for detecting and quantifying modified ribonucleosides with accuracy and precision. However, variations in RNA isolation, processing, and LC-MS/MS analysis have hindered reproducibility across laboratories, which is essential for accurate quantification of RNA modifications. As guidance toward harmonization, we report a multi-laboratory comparison of workflows for LC-MS/MS RNA modification analysis. We compared protocols for sample shipment, RNA hydrolysis, LC-MS/MS analysis, and data processing among three laboratories working with the same total RNA samples. We detected and quantified 17 modifications consistently across protocols and operators, with another 7 that were sensitive to experimental conditions, reagent contamination, and ribonucleoside instability, leading to poor precision among laboratories. Agreement among the three labs was strong, with coefficients of variation of 20% and 10% for relative and absolute quantification, respectively. These findings establish a robust and readily adoptable epitranscriptome analytical platform that enables reliable comparisons across laboratories.
期刊介绍:
Nucleic Acids Research (NAR) is a scientific journal that publishes research on various aspects of nucleic acids and proteins involved in nucleic acid metabolism and interactions. It covers areas such as chemistry and synthetic biology, computational biology, gene regulation, chromatin and epigenetics, genome integrity, repair and replication, genomics, molecular biology, nucleic acid enzymes, RNA, and structural biology. The journal also includes a Survey and Summary section for brief reviews. Additionally, each year, the first issue is dedicated to biological databases, and an issue in July focuses on web-based software resources for the biological community. Nucleic Acids Research is indexed by several services including Abstracts on Hygiene and Communicable Diseases, Animal Breeding Abstracts, Agricultural Engineering Abstracts, Agbiotech News and Information, BIOSIS Previews, CAB Abstracts, and EMBASE.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


