Design, synthesis, and biological evaluation of quinazolin-4-one-based selective HDAC6 inhibitors targeting serine 531, histidine 614 residues, and the L1 and L2 loop
IF 5.9
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
Abstract
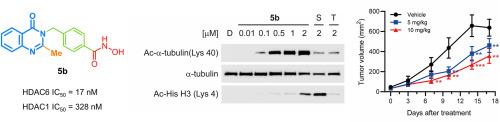
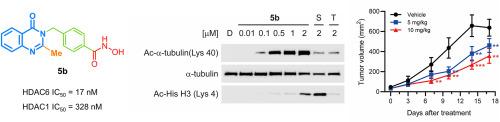
Histone deacetylase 6 (HDAC6) is a crucial therapeutic target for a variety of diseases, including inflammation, autoimmune disorders, neurodegenerative diseases, cancer, viral infections, and drug addiction. Therefore, the discovery of selective HDAC6 inhibitors is essential for clinical application. In this study, we designed and synthesized a novel series of quinazolin-4-one-based selective HDAC6 inhibitors. Among them, the most potent compound, 5b (IC50, 17.15 nM, HDAC6) exhibited 19-fold selectivity over HDAC1 and demonstrated significant anti-proliferative activity, with a GI50 value of 2.4 μM against the MCF-7/ADR cell line. Additionally, compound 5b effectively induced the acetylation of α-tubulin, without affecting histone H3 acetylation in MCF-7/ADR cells, confirming its selectivity toward HDAC6. Compound 5b effectively suppressed cell proliferation by inducing apoptosis, as evidenced by colony formation assays and FACS analysis. Molecular docking study revealed that compound 5b effectively occupied the active site of HDAC6, supporting its strong binding affinity and selectivity. In vitro liver microsomal stability studies revealed that compound 5b was stable in both human and mouse liver microsomes. Furthermore, in an HCT116 xenograft mouse model, compound 5b significantly inhibited tumor growth without affecting body weight. The combination of in vitro and in vivo studies provides robust evidence supporting the potential of compound 5b as a highly potent and selective HDAC6 inhibitor, possessing promising anti-proliferative and apoptosis-inducing properties for further preclinical development.
针对丝氨酸531、组氨酸614残基以及L1和L2环的喹唑啉-4- 1选择性HDAC6抑制剂的设计、合成和生物学评价
组蛋白去乙酰化酶6 (HDAC6)是多种疾病的重要治疗靶点,包括炎症、自身免疫性疾病、神经退行性疾病、癌症、病毒感染和药物成瘾。因此,发现选择性HDAC6抑制剂对临床应用至关重要。在这项研究中,我们设计并合成了一系列新的喹唑啉-4- 1基选择性HDAC6抑制剂。其中,活性最强的化合物5b (IC50为17.15 nM, HDAC6)对MCF-7/ADR细胞株的选择性为HDAC1的19倍,具有显著的抗增殖活性,其GI50值为2.4 μM。此外,化合物5b有效诱导α-微管蛋白乙酰化,而不影响MCF-7/ADR细胞中组蛋白H3乙酰化,证实其对HDAC6的选择性。化合物5b通过诱导细胞凋亡有效抑制细胞增殖,集落形成实验和FACS分析证实了这一点。分子对接研究表明,化合物5b有效占据HDAC6的活性位点,支持其较强的结合亲和力和选择性。体外肝微粒体稳定性研究表明,化合物5b在人和小鼠肝微粒体中都是稳定的。此外,在HCT116异种移植小鼠模型中,化合物5b在不影响体重的情况下显著抑制肿瘤生长。体外和体内研究的结合提供了强有力的证据,支持化合物5b作为一种高效和选择性的HDAC6抑制剂的潜力,具有抗增殖和诱导细胞凋亡的特性,可用于进一步的临床前开发。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


