Gergely Laczkó, , , Imre Pápai*, , and , Péter R. Nagy*,
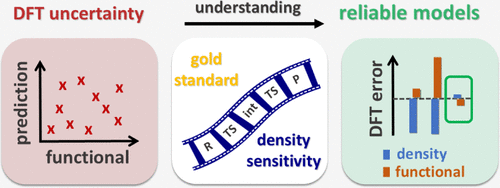
{"title":"Understanding DFT Uncertainties for More Reliable Reactivity Predictions by Advancing the Analysis of Error Sources","authors":"Gergely Laczkó, , , Imre Pápai*, , and , Péter R. Nagy*, ","doi":"10.1021/acs.jctc.5c00985","DOIUrl":null,"url":null,"abstract":"<p >Decades of advancements and thousands of successful applications have contributed to the reliability of density functional theory (DFT) methods. Especially in main group chemistry, DFT predictions tend to be increasingly more reliable. In this study, we deeply analyze unexpected (ca. 8–13 kcal/mol) DFT disagreements obtained for a few organic reactions using only widely adopted, modern, hybrid, and higher-rung DFT methods. To understand the underlying causes, we move beyond conventional statistics-based benchmarks by combining recent advances in DFT error decomposition with affordable gold-standard references. This approach helps to characterize and disentangle multiple functional and density-based error types and enables us to find functional(s) suitable for broad mechanistic studies in all studied examples. The proposed tools are cost-efficient, readily accessible, and easy to integrate into routine thermochemistry workflows. While the focus is on main group reactions, the approach is also applicable to transition metal, bio-, and surface chemistry to assist more predictive reactivity modeling.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9483–9497"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/acs.jctc.5c00985","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00985","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Decades of advancements and thousands of successful applications have contributed to the reliability of density functional theory (DFT) methods. Especially in main group chemistry, DFT predictions tend to be increasingly more reliable. In this study, we deeply analyze unexpected (ca. 8–13 kcal/mol) DFT disagreements obtained for a few organic reactions using only widely adopted, modern, hybrid, and higher-rung DFT methods. To understand the underlying causes, we move beyond conventional statistics-based benchmarks by combining recent advances in DFT error decomposition with affordable gold-standard references. This approach helps to characterize and disentangle multiple functional and density-based error types and enables us to find functional(s) suitable for broad mechanistic studies in all studied examples. The proposed tools are cost-efficient, readily accessible, and easy to integrate into routine thermochemistry workflows. While the focus is on main group reactions, the approach is also applicable to transition metal, bio-, and surface chemistry to assist more predictive reactivity modeling.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


