The critical role of the phosphorylation of STAT3 at Y705 in ALCL-associated NPM-ALK-induced transforming activity
IF 3.7
2区 生物学
Q2 CELL BIOLOGY
引用次数: 0
Abstract
The fusion protein nucleophosmin-anaplastic lymphoma kinase (NPM-ALK) drives oncogenesis in anaplastic large cell lymphoma (ALCL) by activating signal transducer and activator of transcription 3 (STAT3). NPM-ALK requires its kinase activity to induce STAT3 phosphorylation at tyrosine 705 (Y705) and promotes serine 727 (S727) phosphorylation via JNK activation. However, the role of these modifications in NPM-ALK-driven cellular transformation remains unclear. We herein utilized murine Ba/F3 cells expressing NPM-ALK to investigate the impact of these modifications. STAT3 knockdown, followed by reconstitution with wild-type STAT3 or its mutants (Y705F and S727A) revealed that Y705 phosphorylation was essential for NPM-ALK-mediated transformation. STAT3 knockdown suppressed the expression of NPM-ALK-induced STAT3 target genes (c-Myc, Pim, IL-6, and SOCS3) as well as cell proliferation, tumor formation, and spleen, liver, and lymph node enlargement. These effects were restored upon reconstitution with wild-type STAT3 or the S727A mutant, but not the Y705F mutant, confirming the essential role of Y705 phosphorylation in these biological processes. Additionally, wild-type and mutant STAT3 proteins exhibited differential stability, with Y705F being less stable and S727A being more stable. Lysosomal inhibition by bafilomycin A1 increased the expression of wild-type STAT3 and the Y705F mutant, but had no effect on the S727A mutant. Cycloheximide chase assays further confirmed that S727 phosphorylation regulated STAT3 degradation via the lysosomal pathway. These results identify a novel NPM-ALK-induced oncogenic mechanism mediated by STAT3 phosphorylation, highlighting potential therapeutic targets for ALCL.
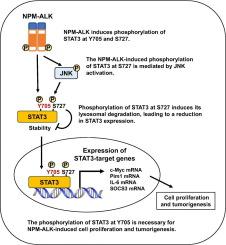
STAT3在Y705位点的磷酸化在alcl相关的npm - alk诱导的转化活性中的关键作用。
融合蛋白核磷蛋白-间变性淋巴瘤激酶(NPM-ALK)通过激活信号转导因子和转录激活因子3 (STAT3)来驱动间变性大细胞淋巴瘤(ALCL)的肿瘤发生。NPM-ALK需要其激酶活性来诱导STAT3酪氨酸705 (Y705)磷酸化,并通过JNK激活促进丝氨酸727 (S727)磷酸化。然而,这些修饰在npm - alk驱动的细胞转化中的作用尚不清楚。我们利用表达NPM-ALK的小鼠Ba/F3细胞来研究这些修饰的影响。STAT3敲除,随后与野生型STAT3或其突变体(Y705F和S727A)重组,表明Y705磷酸化对于npm - alk介导的转化至关重要。STAT3敲低抑制了npm - alk诱导的STAT3靶基因(c-Myc、Pim、IL-6和SOCS3)的表达,以及细胞增殖、肿瘤形成和脾脏、肝脏和淋巴结肿大。这些效应在野生型STAT3或S727A突变体重组后得以恢复,但在Y705F突变体中却没有,这证实了Y705磷酸化在这些生物过程中的重要作用。此外,野生型和突变型STAT3蛋白表现出不同的稳定性,Y705F不太稳定,S727A更稳定。巴菲霉素A1对溶酶体的抑制增加了野生型和Y705F突变体STAT3的表达,但对S727A没有影响。环己亚胺追踪实验进一步证实S727磷酸化通过溶酶体途径调节STAT3降解。这些结果确定了一种新的由STAT3磷酸化介导的npm - alk诱导的致癌机制,突出了ALCL的潜在治疗靶点。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Cellular signalling
生物-细胞生物学
CiteScore
8.40
自引率
0.00%
发文量
250
审稿时长
27 days
期刊介绍:
Cellular Signalling publishes original research describing fundamental and clinical findings on the mechanisms, actions and structural components of cellular signalling systems in vitro and in vivo.
Cellular Signalling aims at full length research papers defining signalling systems ranging from microorganisms to cells, tissues and higher organisms.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


