Ni Zhan, , , William A. Wheeler, , , Gil Goldshlager, , , Elif Ertekin, , , Ryan P. Adams, , and , Lucas K. Wagner*,
{"title":"Expressivity of Determinantal Ansatzes for Neural Network Wave Functions","authors":"Ni Zhan, , , William A. Wheeler, , , Gil Goldshlager, , , Elif Ertekin, , , Ryan P. Adams, , and , Lucas K. Wagner*, ","doi":"10.1021/acs.jctc.5c01243","DOIUrl":null,"url":null,"abstract":"<p >Neural network wave functions have shown promise as a way to achieve high accuracy in solving the many-body quantum problem. These wave functions most commonly use a determinant or a sum of determinants to antisymmetrize many-body orbitals, which are described by a neural network. In many cases, the wave function is projected onto a fixed-spin state. Such a treatment is allowed for spin-independent operators; however, it cannot be applied to spin-dependent problems, such as Hamiltonians containing spin–orbit interactions. We show that for spin-independent Hamiltonians, a strict upper bound property is obeyed between a traditional Hartree–Fock-like determinant, full spinor wave function, the full determinant wave function, and a generalized spinor wave function. The relationship between a spinor wave function and the full determinant arises because the full determinant wave function is the spinor wave function projected onto a fixed-spin, after which antisymmetry is implicitly restored in the spin-independent case. For spin-dependent Hamiltonians, the full determinant wave function is not applicable, because it is not antisymmetric. Numerical experiments on the H<sub>3</sub> molecule and two-dimensional homogeneous electron gas confirm these bounds.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9612–9619"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01243","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
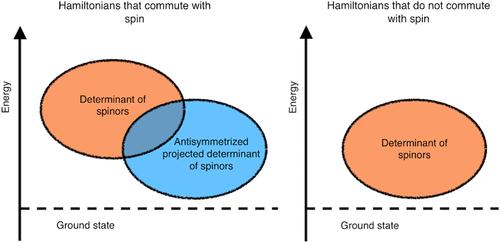
Neural network wave functions have shown promise as a way to achieve high accuracy in solving the many-body quantum problem. These wave functions most commonly use a determinant or a sum of determinants to antisymmetrize many-body orbitals, which are described by a neural network. In many cases, the wave function is projected onto a fixed-spin state. Such a treatment is allowed for spin-independent operators; however, it cannot be applied to spin-dependent problems, such as Hamiltonians containing spin–orbit interactions. We show that for spin-independent Hamiltonians, a strict upper bound property is obeyed between a traditional Hartree–Fock-like determinant, full spinor wave function, the full determinant wave function, and a generalized spinor wave function. The relationship between a spinor wave function and the full determinant arises because the full determinant wave function is the spinor wave function projected onto a fixed-spin, after which antisymmetry is implicitly restored in the spin-independent case. For spin-dependent Hamiltonians, the full determinant wave function is not applicable, because it is not antisymmetric. Numerical experiments on the H3 molecule and two-dimensional homogeneous electron gas confirm these bounds.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


