Tonghuan Jiang, , , Nikolay A. Bogdanov*, , , Ali Alavi*, , and , Ji Chen*,
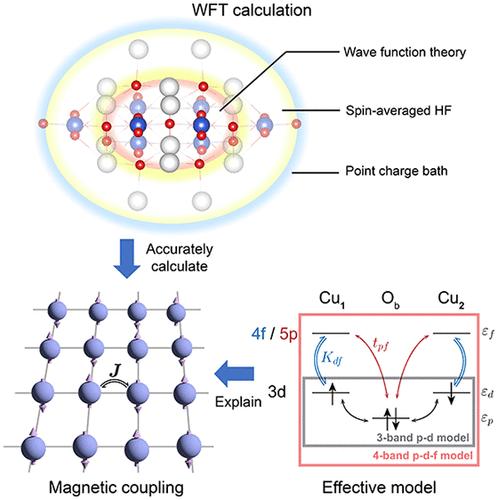
{"title":"Individual and Cooperative Superexchange Enhancement in Cuprates","authors":"Tonghuan Jiang, , , Nikolay A. Bogdanov*, , , Ali Alavi*, , and , Ji Chen*, ","doi":"10.1021/acs.jctc.5c00755","DOIUrl":null,"url":null,"abstract":"<p >It is now widely accepted that the antiferromagnetic coupling within high-temperature superconductors strongly exhibits a profound correlation with the upper limit of the superconducting transition temperature these materials can reach. Thus, accurately calculating the positive and negative mechanisms that influence magnetic coupling in specific materials is crucial for the exploration of superconductivity at higher temperatures. Nevertheless, it is notoriously difficult to establish a complete description of electron correlations employing ab initio theories because of the large number of orbitals involved. In this study, we tackle the challenge of achieving high-level ab initio wave function theory calculations that allow an explicit treatment of electron correlations associated with a large number of high-energy orbitals. We elucidate the atomic-shell-wise contributions to the superexchange coupling in the lanthanum cuprate, including individual effects of high-energy orbitals (Cu 4d, 5d, 4f, 5p) and cooperative effects between the core and these high-energy orbitals. Specifically, the prominent contributions from Cu 4d, 5d, 4f, and 5p give rise to a rich collection of previously unexamined superexchange channels. We propose a <i>p</i>-<i>d</i>-<i>f</i> model to universally account for the contributions of high-energy orbitals at copper sites. Our calculations and physical rationalizations offer a more robust theoretical foundation for investigating cuprate-type high-temperature superconductors.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9364–9375"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00755","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
It is now widely accepted that the antiferromagnetic coupling within high-temperature superconductors strongly exhibits a profound correlation with the upper limit of the superconducting transition temperature these materials can reach. Thus, accurately calculating the positive and negative mechanisms that influence magnetic coupling in specific materials is crucial for the exploration of superconductivity at higher temperatures. Nevertheless, it is notoriously difficult to establish a complete description of electron correlations employing ab initio theories because of the large number of orbitals involved. In this study, we tackle the challenge of achieving high-level ab initio wave function theory calculations that allow an explicit treatment of electron correlations associated with a large number of high-energy orbitals. We elucidate the atomic-shell-wise contributions to the superexchange coupling in the lanthanum cuprate, including individual effects of high-energy orbitals (Cu 4d, 5d, 4f, 5p) and cooperative effects between the core and these high-energy orbitals. Specifically, the prominent contributions from Cu 4d, 5d, 4f, and 5p give rise to a rich collection of previously unexamined superexchange channels. We propose a p-d-f model to universally account for the contributions of high-energy orbitals at copper sites. Our calculations and physical rationalizations offer a more robust theoretical foundation for investigating cuprate-type high-temperature superconductors.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


