{"title":"A Generalized Solid Solution Framework for the Gibbs Free Energy Calculation","authors":"Yang Huang, and , Jingrun Chen*, ","doi":"10.1021/acs.jctc.5c00524","DOIUrl":null,"url":null,"abstract":"<p >We propose a generalized solid solution model for calculating configurational contribution to the Gibbs free energy at finite temperatures, incorporating a crystal graph-based on-site energy approach. By leveraging linear graph neural networks, our method unifies pair-based and cluster expansion approaches, enabling broad applicability across crystal structures. Fractional occupation is physically interpreted via mean-field theory, while entropy is modeled using ideal mixing with extended site constraints. To resolve the constants of compositions, we implement three key strategies. First, we employ a softmax-based variable transformation. Second, we introduce a gradient projection method that preserves species composition throughout the optimization process by constraining updates within a subspace that maintains the desired elemental ratios. Finally, a renormalization step is incorporated to correct numerical deviations, ensuring strict adherence to the target composition. We then apply our model to the Mo–Nb–Ta-W quaternary system, achieving an energy model MAE of 1.24 meV. Predicted phase transition temperatures for equal atomic binary alloys align well with expectations, identifying phase separation in MoNb and order–disorder transitions in MoTa, MoW, TaW, NbTa, and NbW. At low temperatures, stable configurations lie below the convex hull of the training data set, demonstrating the model’s predictive accuracy. Further analysis of MoNbTaW reveals transition temperatures at 950 and 400 K, with observed asymmetry in Mo/W sublattices. Finally, we extend our approach to ternary phase diagram predictions using Gibbs free energy interpolation and second-derivative analysis, yielding phase diagrams in agreement with optimized atomic configurations.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9982–9992"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00524","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
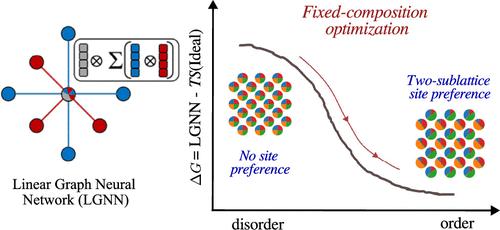
We propose a generalized solid solution model for calculating configurational contribution to the Gibbs free energy at finite temperatures, incorporating a crystal graph-based on-site energy approach. By leveraging linear graph neural networks, our method unifies pair-based and cluster expansion approaches, enabling broad applicability across crystal structures. Fractional occupation is physically interpreted via mean-field theory, while entropy is modeled using ideal mixing with extended site constraints. To resolve the constants of compositions, we implement three key strategies. First, we employ a softmax-based variable transformation. Second, we introduce a gradient projection method that preserves species composition throughout the optimization process by constraining updates within a subspace that maintains the desired elemental ratios. Finally, a renormalization step is incorporated to correct numerical deviations, ensuring strict adherence to the target composition. We then apply our model to the Mo–Nb–Ta-W quaternary system, achieving an energy model MAE of 1.24 meV. Predicted phase transition temperatures for equal atomic binary alloys align well with expectations, identifying phase separation in MoNb and order–disorder transitions in MoTa, MoW, TaW, NbTa, and NbW. At low temperatures, stable configurations lie below the convex hull of the training data set, demonstrating the model’s predictive accuracy. Further analysis of MoNbTaW reveals transition temperatures at 950 and 400 K, with observed asymmetry in Mo/W sublattices. Finally, we extend our approach to ternary phase diagram predictions using Gibbs free energy interpolation and second-derivative analysis, yielding phase diagrams in agreement with optimized atomic configurations.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


