Luigi Crisci*, , , Federico Lazzari, , and , Vincenzo Barone*,
{"title":"Accurate, Affordable and Unsupervised: Analytical F12 Gradients Driven by Generalized Internal Coordinates","authors":"Luigi Crisci*, , , Federico Lazzari, , and , Vincenzo Barone*, ","doi":"10.1021/acs.jpclett.5c02292","DOIUrl":null,"url":null,"abstract":"<p >Accurate yet affordable quantum-chemical predictions are essential in several fields of molecular sciences, such as high-resolution rotational and vibrational spectroscopy. Despite major methodological and technological advances, benchmark-level methods remain prohibitive for molecules of realistic size. We present a modular implementation of the Pisa Composite Schemes (PCS), where analytical gradients ensure robust optimizations and efficient frequency calculations. PCS variants are combined hierarchically to reduce the iterations required in geometry optimizations with the most accurate and costly model, while multilayer ONIOM descriptions extend applicability by treating chemically central regions at high accuracy and peripheral fragments with lower-cost DFT variants. This strategy achieves near-spectroscopic accuracy for medium-sized molecules, including challenging cases such as CN-substituted PAHs and transition states. Freely available and user-friendly, the platform paves the way for routine, spectroscopically accurate simulations of complex systems with quantified uncertainty.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"16 38","pages":"9985–9992"},"PeriodicalIF":4.6000,"publicationDate":"2025-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.5c02292","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
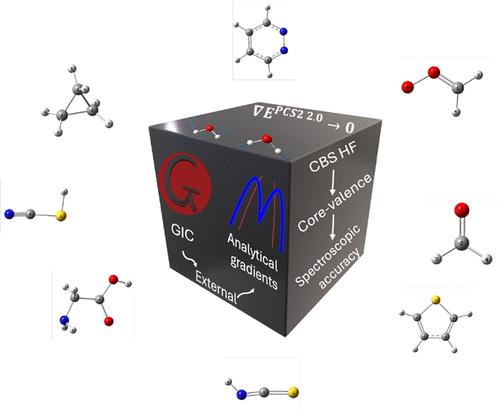
Accurate yet affordable quantum-chemical predictions are essential in several fields of molecular sciences, such as high-resolution rotational and vibrational spectroscopy. Despite major methodological and technological advances, benchmark-level methods remain prohibitive for molecules of realistic size. We present a modular implementation of the Pisa Composite Schemes (PCS), where analytical gradients ensure robust optimizations and efficient frequency calculations. PCS variants are combined hierarchically to reduce the iterations required in geometry optimizations with the most accurate and costly model, while multilayer ONIOM descriptions extend applicability by treating chemically central regions at high accuracy and peripheral fragments with lower-cost DFT variants. This strategy achieves near-spectroscopic accuracy for medium-sized molecules, including challenging cases such as CN-substituted PAHs and transition states. Freely available and user-friendly, the platform paves the way for routine, spectroscopically accurate simulations of complex systems with quantified uncertainty.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


