{"title":"Accelerated Bayesian inference of population size history from recombining sequence data","authors":"Jonathan Terhorst","doi":"10.1038/s41588-025-02323-x","DOIUrl":null,"url":null,"abstract":"<p>This study introduces population history learning by averaging sampled histories (PHLASH), a new method for inferring population history from whole-genome sequence data. It works by drawing random, low-dimensional projections of the coalescent intensity function from the posterior distribution of a pairwise sequentially Markovian coalescent-like model and averaging them together to form an accurate and adaptive estimator. On simulated data, PHLASH tends to be faster and have lower error than several competing methods, including SMC++, MSMC2 and FITCOAL. Moreover, it provides automatic uncertainty quantification and leads to new Bayesian testing procedures for detecting population structure and ancient bottlenecks. The key technical advance is a new algorithm for computing the score function (gradient of the log likelihood) of a coalescent hidden Markov model, which has the same computational cost as evaluating the log likelihood. PHLASH has been released as an easy-to-use Python software package and leverages graphics processing unit acceleration when available.</p>","PeriodicalId":18985,"journal":{"name":"Nature genetics","volume":"28 1","pages":""},"PeriodicalIF":29.0000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41588-025-02323-x","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
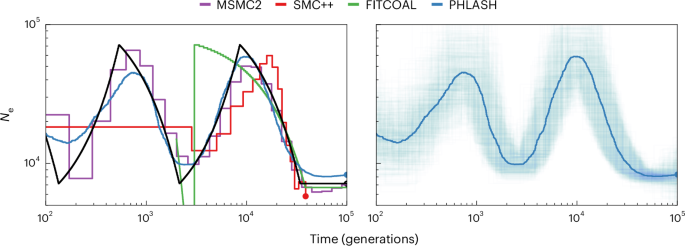
This study introduces population history learning by averaging sampled histories (PHLASH), a new method for inferring population history from whole-genome sequence data. It works by drawing random, low-dimensional projections of the coalescent intensity function from the posterior distribution of a pairwise sequentially Markovian coalescent-like model and averaging them together to form an accurate and adaptive estimator. On simulated data, PHLASH tends to be faster and have lower error than several competing methods, including SMC++, MSMC2 and FITCOAL. Moreover, it provides automatic uncertainty quantification and leads to new Bayesian testing procedures for detecting population structure and ancient bottlenecks. The key technical advance is a new algorithm for computing the score function (gradient of the log likelihood) of a coalescent hidden Markov model, which has the same computational cost as evaluating the log likelihood. PHLASH has been released as an easy-to-use Python software package and leverages graphics processing unit acceleration when available.
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


