Hengyuan Shen, , , Nicola Bogo, , , Christopher J. Stein, , and , Martin Head-Gordon*,
{"title":"Understanding Electronic Excitations Between Single Determinants with Occupied-Virtual Orbitals for Chemical Valence","authors":"Hengyuan Shen, , , Nicola Bogo, , , Christopher J. Stein, , and , Martin Head-Gordon*, ","doi":"10.1021/acs.jctc.5c01029","DOIUrl":null,"url":null,"abstract":"<p >One approach to calculating electronic excited states treats both ground and excited states as single determinants, either by direct optimization or with the aid of constraints. In this work, we extend the theory of occupied-virtual orbitals for chemical valence (OVOCV) to analyze the orbital character of excitations computed in this way. An intermediate frozen state that is polarization-free is introduced to cleanly separate the primary excitation from the accompanying orbital relaxation of spectator orbitals. A variety of chemical examples are reported using the OVOCV excitation analysis on orbital-optimized density functional theory (OO–DFT) calculations, including charge-transfer excitations, core excitations and singly and doubly excited valence states. Orbital relaxation effects are typically collective, and can be as large as 4–5 eV (with roughly 0.1 <i>e</i><sup>–</sup> promoted) in charge transfer states, and even larger in core excited states. OVOCV analysis differs from natural transition orbital (NTO) analysis; we show that direct use of NTOs can largely obscure the role of orbital relaxation in favor of the primary excitation.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9525–9537"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01029","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
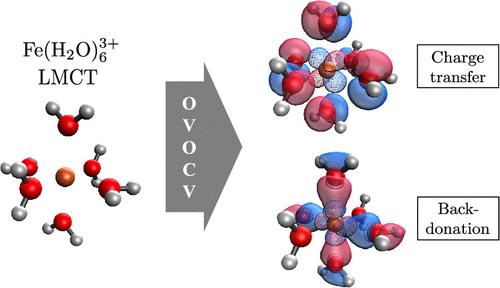
One approach to calculating electronic excited states treats both ground and excited states as single determinants, either by direct optimization or with the aid of constraints. In this work, we extend the theory of occupied-virtual orbitals for chemical valence (OVOCV) to analyze the orbital character of excitations computed in this way. An intermediate frozen state that is polarization-free is introduced to cleanly separate the primary excitation from the accompanying orbital relaxation of spectator orbitals. A variety of chemical examples are reported using the OVOCV excitation analysis on orbital-optimized density functional theory (OO–DFT) calculations, including charge-transfer excitations, core excitations and singly and doubly excited valence states. Orbital relaxation effects are typically collective, and can be as large as 4–5 eV (with roughly 0.1 e– promoted) in charge transfer states, and even larger in core excited states. OVOCV analysis differs from natural transition orbital (NTO) analysis; we show that direct use of NTOs can largely obscure the role of orbital relaxation in favor of the primary excitation.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


