SAMHD1 promotes cardiac repair post myocardial infarction by targeting NR4a1 to regulate macrophage metabolic reprogramming
IF 13
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
Abstract
Introduction
Myocardial infarction (MI) is one of the leading causes of high mortality worldwide. Accumulating evidence suggests that macrophages emerge as the predominant immune population within the post-MI cardiac environment, serving as critical modulators that coordinate inflammatory cascades during myocardial repair.Objectives
The main objective of this study was to explore the effects of sterile alpha motif and HD domain-containing protein 1 (SAMHD1) on myocardial remodeling post-MI and to elucidate its potential mechanism.We used MI mouse model ligation of the left anterior descending coronary artery (LAD) to investigate the role of SAMHD1 in MI. To assess the role of SAMHD1 in MI, we generated both macrophage-specific knockout and overexpression mice. To investigate the mechanisms by which SAMHD1 regulates MI progression, we employed transcriptomics sequencing and nontargeted metabolomics.Results
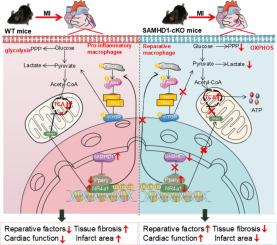
SAMHD1 was significantly upregulated in mouse cardiac macrophages on day 3 post-MI and was closely associated with immune responses. We found that SAMHD1 deficiency facilitated myocardial repair. We found that SAMHD1 deficiency confers cardioprotection through metabolic reprogramming mechanisms: increased mitochondrial oxidative phosphorylation capacity coupled with increased production of the anti-inflammatory metabolite itaconic acid and suppression of the pentose phosphate pathway and lactate biosynthesis. We found that these metabolic shifts facilitated macrophage differentiation by promoting a transition toward reparative macrophage populations. Furthermore, SAMHD1 deficiency drives macrophage phenotypic switching through the transcriptional suppression of NR4a1. More importantly, we have shown that SAMHD1 deficiency promotes the interaction between NR4a1 and Pparγ, which facilitates NR4a1 ubiquitination-dependent degradation.Conclusion
Our study revealed that macrophage-specific SAMHD1 deletion confers post-MI cardioprotection. More importantly, we demonstrated that NR4a1, a downstream target of SAMHD1, mediates the cardioprotective effects of SAMHD1 deficiency post-MI by regulating the remodeling of macrophage energy metabolism to promote the macrophage reparative phenotype.
SAMHD1通过靶向NR4a1调控巨噬细胞代谢重编程,促进心肌梗死后心脏修复
心肌梗死(MI)是全球高死亡率的主要原因之一。越来越多的证据表明,巨噬细胞在心肌梗死后的心脏环境中作为主要的免疫群体出现,在心肌修复过程中作为协调炎症级联反应的关键调节剂。目的探讨无菌α基序和含HD结构域蛋白1 (SAMHD1)在心肌梗死后心肌重构中的作用,并探讨其潜在机制。我们使用结扎左冠状动脉前降支(LAD)的心肌梗死小鼠模型来研究SAMHD1在心肌梗死中的作用。为了评估SAMHD1在心肌梗死中的作用,我们制造了巨噬细胞特异性敲除和过表达小鼠。为了研究SAMHD1调控心肌梗死进展的机制,我们采用了转录组学测序和非靶向代谢组学。结果心肌梗死后第3天,小鼠心脏巨噬细胞中samhd1表达显著上调,并与免疫反应密切相关。我们发现SAMHD1缺失促进心肌修复。我们发现SAMHD1缺陷通过代谢重编程机制赋予心脏保护作用:线粒体氧化磷酸化能力增加,抗炎代谢物衣康酸的产生增加,戊糖磷酸途径和乳酸生物合成受到抑制。我们发现,这些代谢变化通过促进巨噬细胞向修复性巨噬细胞群体的过渡,促进了巨噬细胞的分化。此外,SAMHD1缺陷通过抑制NR4a1转录驱动巨噬细胞表型转换。更重要的是,我们已经证明SAMHD1缺陷促进了NR4a1和Pparγ之间的相互作用,这促进了NR4a1泛素化依赖性降解。我们的研究表明,巨噬细胞特异性SAMHD1缺失具有心肌梗死后的心脏保护作用。更重要的是,我们证明了SAMHD1的下游靶点NR4a1通过调节巨噬细胞能量代谢的重塑,促进巨噬细胞的修复表型,介导心肌梗死后SAMHD1缺乏的心脏保护作用。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Advanced Research
Multidisciplinary-Multidisciplinary
CiteScore
21.60
自引率
0.90%
发文量
280
审稿时长
12 weeks
期刊介绍:
Journal of Advanced Research (J. Adv. Res.) is an applied/natural sciences, peer-reviewed journal that focuses on interdisciplinary research. The journal aims to contribute to applied research and knowledge worldwide through the publication of original and high-quality research articles in the fields of Medicine, Pharmaceutical Sciences, Dentistry, Physical Therapy, Veterinary Medicine, and Basic and Biological Sciences.
The following abstracting and indexing services cover the Journal of Advanced Research: PubMed/Medline, Essential Science Indicators, Web of Science, Scopus, PubMed Central, PubMed, Science Citation Index Expanded, Directory of Open Access Journals (DOAJ), and INSPEC.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


