{"title":"Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 deficiency with severe hyperglycemia in a child: A rare case report.","authors":"Chang Dong, Tiantian Lu, Yazhou Jiang, Zihao Yan, Suyue Zhu","doi":"10.1177/03000605251375537","DOIUrl":null,"url":null,"abstract":"<p><p>Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 (HMGCS2) deficiency is an exceptionally rare autosomal recessive metabolic disorder that impairs ketogenesis. It is typically characterized by hypoketotic hypoglycemia during periods of fasting or metabolic stress. Notably, severe hyperglycemia as an initial presenting symptom has not been previously reported. We report the case of a 6-month-old girl who suddenly developed coma after 1 day of fasting due to repeated vomiting during pneumonia. At presentation, she had hyperglycemia (25.8 mmol/L), ketonuria (1+), glucosuria (3+), metabolic acidosis (pH 6.90), elevated serum alanine transaminase and aspartate aminotransferase levels, increased blood ammonia levels, and liver enlargement on ultrasound. However, fasting insulin, glucagon, and glycated hemoglobin levels were all within the normal range. Whole-exome sequencing identified compound heterozygous mutations in the HMGCS2 gene-c.1175C>T (p.S392L) inherited from the father and c.719A>T (p.A240V) inherited from the mother-thereby confirming the diagnosis of HMGCS2 deficiency. This case highlights severe hyperglycemia as an atypical clinical feature of HMGCS2 deficiency. Increased awareness of such rare manifestations may assist in improving early diagnosis and treatment of this condition.</p>","PeriodicalId":16129,"journal":{"name":"Journal of International Medical Research","volume":"53 9","pages":"3000605251375537"},"PeriodicalIF":1.5000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12432301/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of International Medical Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1177/03000605251375537","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/9/12 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
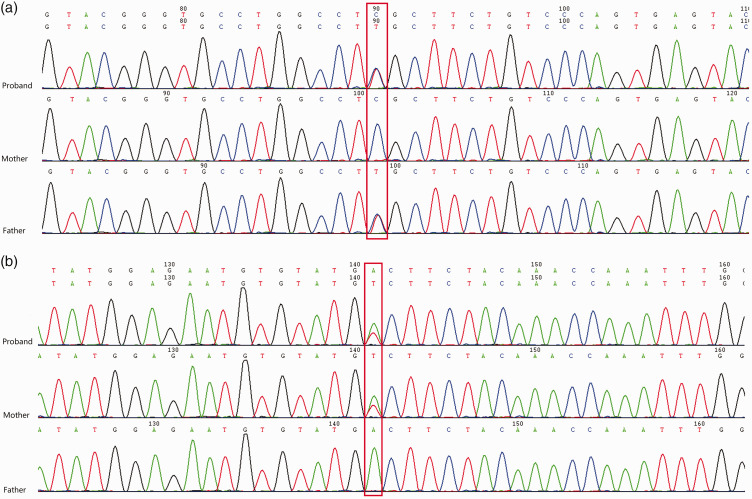
Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 (HMGCS2) deficiency is an exceptionally rare autosomal recessive metabolic disorder that impairs ketogenesis. It is typically characterized by hypoketotic hypoglycemia during periods of fasting or metabolic stress. Notably, severe hyperglycemia as an initial presenting symptom has not been previously reported. We report the case of a 6-month-old girl who suddenly developed coma after 1 day of fasting due to repeated vomiting during pneumonia. At presentation, she had hyperglycemia (25.8 mmol/L), ketonuria (1+), glucosuria (3+), metabolic acidosis (pH 6.90), elevated serum alanine transaminase and aspartate aminotransferase levels, increased blood ammonia levels, and liver enlargement on ultrasound. However, fasting insulin, glucagon, and glycated hemoglobin levels were all within the normal range. Whole-exome sequencing identified compound heterozygous mutations in the HMGCS2 gene-c.1175C>T (p.S392L) inherited from the father and c.719A>T (p.A240V) inherited from the mother-thereby confirming the diagnosis of HMGCS2 deficiency. This case highlights severe hyperglycemia as an atypical clinical feature of HMGCS2 deficiency. Increased awareness of such rare manifestations may assist in improving early diagnosis and treatment of this condition.
期刊介绍:
_Journal of International Medical Research_ is a leading international journal for rapid publication of original medical, pre-clinical and clinical research, reviews, preliminary and pilot studies on a page charge basis.
As a service to authors, every article accepted by peer review will be given a full technical edit to make papers as accessible and readable to the international medical community as rapidly as possible.
Once the technical edit queries have been answered to the satisfaction of the journal, the paper will be published and made available freely to everyone under a creative commons licence.
Symposium proceedings, summaries of presentations or collections of medical, pre-clinical or clinical data on a specific topic are welcome for publication as supplements.
Print ISSN: 0300-0605

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


