Three-dimensional Quantitative Structure-activity Relationship, Molecular Docking and Absorption, Distribution, Metabolism, and Excretion Studies of Lidocaine Analogs Pertaining to Voltage-gated Sodium Channel Nav1.7 Inhibition for the Management of Neuropathic Pain.
{"title":"Three-dimensional Quantitative Structure-activity Relationship, Molecular Docking and Absorption, Distribution, Metabolism, and Excretion Studies of Lidocaine Analogs Pertaining to Voltage-gated Sodium Channel Na<sub>v</sub>1.7 Inhibition for the Management of Neuropathic Pain.","authors":"Shiwani Sharma, Priyanka Rana, Neelima Dhingra, Tanzeer Kaur","doi":"10.4103/ijabmr.ijabmr_347_24","DOIUrl":null,"url":null,"abstract":"<p><strong>Aim: </strong>This study aims to design and develop novel lidocaine analogs specific for the Na<sub>v</sub>1.7 channel using <i>in silico</i> approaches.</p><p><strong>Background: </strong>Neuropathic pain (NP) is defined as chronic pain originating from abnormalities found within the nervous system. Voltage-gated sodium channels play a significant role in enhancing neuronal excitability, thus gained significance as a crucial target for developing drugs to treat NP. It consists of 9 different isoforms, with Na<sub>v</sub>1.7 predominantly found in the dorsal root ganglion, playing a crucial role in the pathophysiology of NP. The selective inhibitors targeting the Na<sub>v</sub>1.7 channel hold greater potential for treating NP while minimizing interference with the physiological functions of other sodium channel isoforms.</p><p><strong>Methods: </strong>Atom and field-based three-dimensional (3D) quantitative structure-activity relationship (QSAR) was created using lidocaine analogs to identify the structural features required for the Na<sub>v</sub>1.7 inhibitory activities. Further, the molecular interaction of the scaffold with the Na<sub>v</sub>1.7 channel VSD4 was studied by docking the molecules with it followed by absorption, distribution, metabolism, and excretion (ADME) analysis.</p><p><strong>Results: </strong>The 3D QSAR studies revealed that the presence of hydrophobic groups and steric parameters heightened the specificity for Na<sub>v</sub>1.7 channel. Docking analysis revealed that 4 compounds, i.e., A15, A14, A6, and A5, exhibited the highest binding affinity in comparison to reference drug lidocaine. Furthermore, ADME predictions indicated that the compounds exhibited favorable characteristics in terms of oral bioavailability and solubility.</p><p><strong>Conclusion: </strong>This research offers valuable structural insights to improve the specific inhibition of the Na<sub>v</sub>1.7 channel, facilitating the design and development of novel, Na<sub>v</sub>1.7 channel-specific inhibitors.</p>","PeriodicalId":13727,"journal":{"name":"International Journal of Applied and Basic Medical Research","volume":"15 3","pages":"143-151"},"PeriodicalIF":0.8000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12422546/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Applied and Basic Medical Research","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4103/ijabmr.ijabmr_347_24","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/20 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
Aim: This study aims to design and develop novel lidocaine analogs specific for the Nav1.7 channel using in silico approaches.
Background: Neuropathic pain (NP) is defined as chronic pain originating from abnormalities found within the nervous system. Voltage-gated sodium channels play a significant role in enhancing neuronal excitability, thus gained significance as a crucial target for developing drugs to treat NP. It consists of 9 different isoforms, with Nav1.7 predominantly found in the dorsal root ganglion, playing a crucial role in the pathophysiology of NP. The selective inhibitors targeting the Nav1.7 channel hold greater potential for treating NP while minimizing interference with the physiological functions of other sodium channel isoforms.
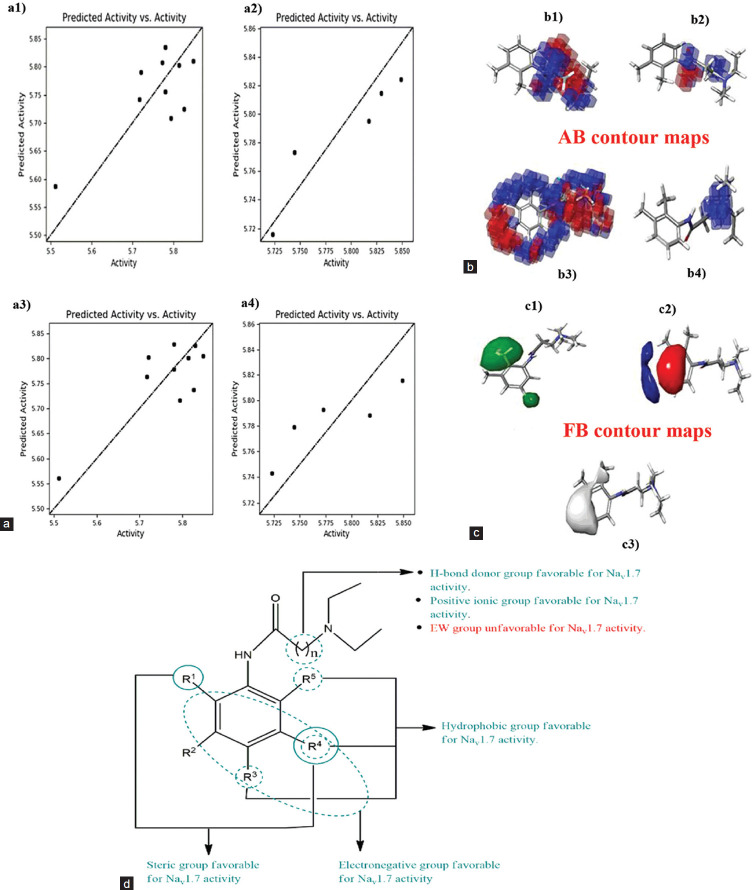
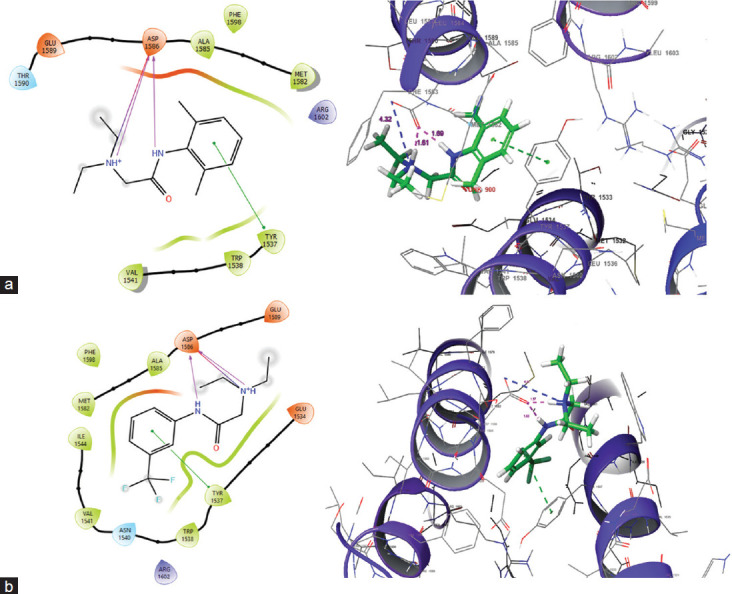
Methods: Atom and field-based three-dimensional (3D) quantitative structure-activity relationship (QSAR) was created using lidocaine analogs to identify the structural features required for the Nav1.7 inhibitory activities. Further, the molecular interaction of the scaffold with the Nav1.7 channel VSD4 was studied by docking the molecules with it followed by absorption, distribution, metabolism, and excretion (ADME) analysis.
Results: The 3D QSAR studies revealed that the presence of hydrophobic groups and steric parameters heightened the specificity for Nav1.7 channel. Docking analysis revealed that 4 compounds, i.e., A15, A14, A6, and A5, exhibited the highest binding affinity in comparison to reference drug lidocaine. Furthermore, ADME predictions indicated that the compounds exhibited favorable characteristics in terms of oral bioavailability and solubility.
Conclusion: This research offers valuable structural insights to improve the specific inhibition of the Nav1.7 channel, facilitating the design and development of novel, Nav1.7 channel-specific inhibitors.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


