DNA methylation cooperates with genomic alterations during non-small cell lung cancer evolution
IF 29
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
Abstract
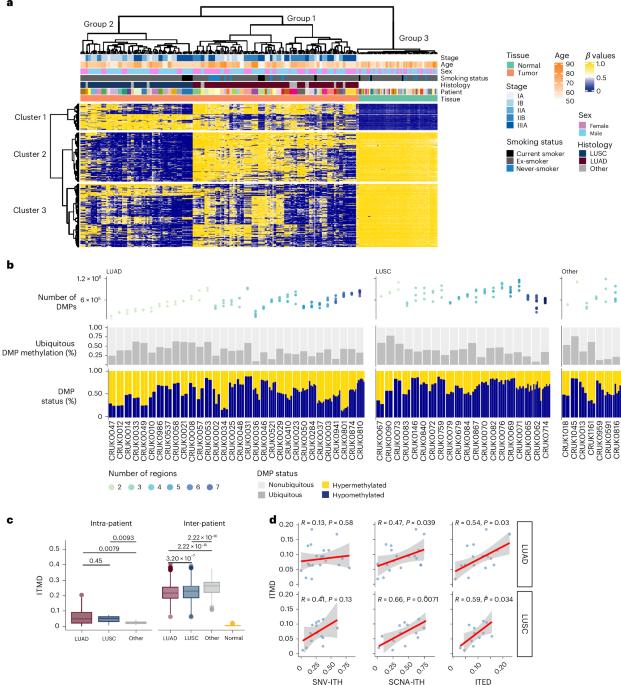
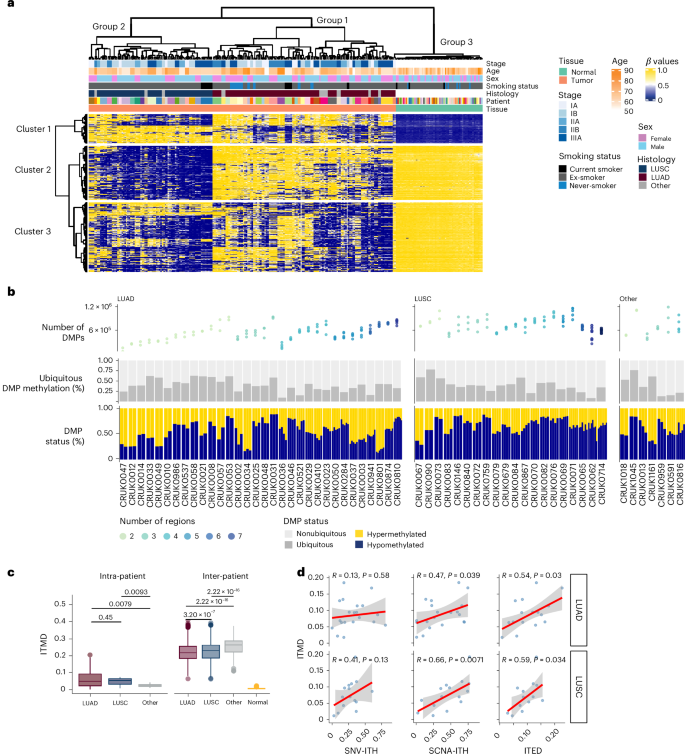
Aberrant DNA methylation has been described in nearly all human cancers, yet its interplay with genomic alterations during tumor evolution is poorly understood. To explore this, we performed reduced representation bisulfite sequencing on 217 tumor and matched normal regions from 59 patients with non-small cell lung cancer from the TRACERx study to deconvolve tumor methylation. We developed two metrics for integrative evolutionary analysis with DNA and RNA sequencing data. Intratumoral methylation distance quantifies intratumor DNA methylation heterogeneity. MR/MN classifies genes based on the rate of hypermethylation at regulatory (MR) versus nonregulatory (MN) CpGs to identify driver genes exhibiting recurrent functional hypermethylation. We identified DNA methylation-linked dosage compensation of essential genes co-amplified with neighboring oncogenes. We propose two complementary mechanisms that converge for copy number alteration-affected chromatin to undergo the epigenetic equivalent of an allosteric activity transition. Hypermethylated driver genes under positive selection may open avenues for therapeutic stratification of patients. Integrated multi-omic analyses using samples from the TRACERx study highlight cross-talk between DNA hypermethylation and genomic lesions in non-small cell lung cancer.
在非小细胞肺癌的进化过程中,DNA甲基化与基因组改变密切相关
几乎在所有人类癌症中都有异常DNA甲基化的描述,但其与肿瘤进化过程中基因组改变的相互作用却知之甚少。为了探讨这一点,我们对来自TRACERx研究的59名非小细胞肺癌患者的217个肿瘤和匹配的正常区域进行了减少代表性的亚硫酸氢盐测序,以反卷积肿瘤甲基化。我们利用DNA和RNA测序数据开发了两个综合进化分析指标。肿瘤内甲基化距离量化肿瘤内DNA甲基化异质性。MR/MN根据调节性(MR)和非调节性(MN) CpGs的高甲基化率对基因进行分类,以确定表现出复发性功能性高甲基化的驱动基因。我们确定了与邻近癌基因共同扩增的必要基因的DNA甲基化相关剂量补偿。我们提出了两种互补的机制,这些机制收敛于拷贝数改变影响的染色质,以经历表观遗传等效的变构活性转变。正选择下的高甲基化驱动基因可能为患者的治疗分层开辟道路。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


