KMT2A-rearranged leukemia: from mechanism to drug development
IF 2.1
4区 医学
Q2 HEMATOLOGY
引用次数: 0
Abstract
Gene rearrangements of the human mixed lineage leukemia (MLL) gene (also known as KMT2A) generate multiple fusion oncoproteins, which cause leukemia with poor prognosis. MLL is an epigenetic regulator that reads and writes epigenetic information and has an evolutionarily conserved role in maintaining expression of Homeotic (HOX) genes during embryonic development. Most MLL gene rearrangements found in leukemia generate a constitutively active version of the wild-type protein, which causes overexpression of HOX and other genes and leukemic transformation of normal hematopoietic progenitors. Elucidating the molecular mechanisms underlying how MLL activates gene expression and how gene rearrangements affect this gene-regulating activity provided therapeutic opportunities to block fusion oncoprotein-specific activities. One uniform molecular dependency of MLL fusion oncoproteins is its interaction with the chromatin-binding partner MENIN that is essential to maintain leukemic transformation. MENIN inhibitors that interfere with the MLL–MENIN interaction have been developed and are now entering clinical practice. Also, the MLL complex physically interacts with several histone acetyl transferases (HATs), including MOZ/MORF, HBO1, and EP300/CREBBP to effect MLL–MENIN-dependent gene activation. Aberrant recruitment of these HATs and other transcriptional effector complexes are key differences between MLL and MLL fusion oncoproteins. In this review, we first summarized our current understanding of wild-type MLL function and the aberrant function of its oncogenic variants. We then discussed in detail how chromosomal translocations generate constitutive-active forms of MLL and categorize them into five major classes. We touched on the collaborative gene activation by MLL and specific interacting HATs. Lastly, we discussed how these mechanistic insights have led to the development of the first-in-class MENIN inhibitors and discussed efforts to anticipate and treat both genetic and nongenetic mechanisms of resistance.
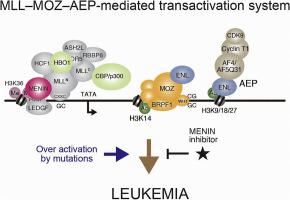
kmt2a重排白血病:从机制到药物开发。
人类MLL基因(也称为KMT2A)的基因重排产生多种融合癌蛋白,导致预后不良的白血病。MLL是一种表观遗传调控因子,具有读取和写入表观遗传信息的功能,在胚胎发育过程中具有维持同源(HOX)基因表达的进化保守作用。在白血病中发现的大多数MLL基因重排产生了野生型蛋白的组成型活性版本,这导致HOX和其他基因的过度表达以及正常造血祖细胞的白血病转化。阐明MLL激活基因表达的分子机制以及基因重排如何影响这种基因调节活性,为阻断融合肿瘤蛋白特异性活性提供了治疗机会。MLL融合癌蛋白的一个统一的分子依赖性是它与染色质结合伙伴MENIN的相互作用,这是维持白血病转化所必需的。干扰MLL-MENIN相互作用的MENIN抑制剂已经开发出来,现在正在进入临床实践。此外,MLL复合体与多种组蛋白乙酰转移酶(HATs)相互作用,包括MOZ/MORF、HBO1和EP300/CREBBP,以影响MLL- menin依赖的基因激活。这些hat和其他转录效应复合物的异常募集是MLL和MLL融合癌蛋白之间的关键区别。在这篇综述中,我们首先总结了我们目前对野生型MLL功能及其致癌变异的异常功能的了解。然后,我们详细讨论了染色体易位如何产生MLL的组成活性形式,并将其分为五大类。我们讨论了MLL和特异性相互作用的hat的协同基因激活。最后,我们讨论了这些机制见解如何导致一流MENIN抑制剂的开发,并讨论了预测和治疗遗传和非遗传耐药机制的努力。摘要:MLL融合癌蛋白通常在不需要额外突变的情况下转化处于特定发育状态的细胞。通过研究这一过程,我们获得了丰富的机制和生物学见解。在这里,我们回顾了导致白血病发展的一些详细的分子机制,以及如何理解这些机制导致靶向治疗的发展,以及正在进行的预测和预防靶向治疗耐药性的努力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Experimental hematology
医学-血液学
CiteScore
5.30
自引率
0.00%
发文量
84
审稿时长
58 days
期刊介绍:
Experimental Hematology publishes new findings, methodologies, reviews and perspectives in all areas of hematology and immune cell formation on a monthly basis that may include Special Issues on particular topics of current interest. The overall goal is to report new insights into how normal blood cells are produced, how their production is normally regulated, mechanisms that contribute to hematological diseases and new approaches to their treatment. Specific topics may include relevant developmental and aging processes, stem cell biology, analyses of intrinsic and extrinsic regulatory mechanisms, in vitro behavior of primary cells, clonal tracking, molecular and omics analyses, metabolism, epigenetics, bioengineering approaches, studies in model organisms, novel clinical observations, transplantation biology and new therapeutic avenues.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


