Machine learning-assisted computational screening of high-entropy alloy catalysts for HCOOH decomposition
IF 9.5
2区 材料科学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Catalytic activities have been found to correlate with the adsorption energies of key intermediates in many catalytic reactions, and computational screening based on the adsorption energies of such intermediates has been widely used for catalyst discovery. High-entropy alloys (HEAs) offer an expansive configuration space, leading to a near-continuous distribution of adsorption energies for intermediates, thereby facilitating the identification of promising catalysts with optimal adsorption energies. However, comprehensive DFT calculations of adsorption energies on HEAs are hindered by the vast number of surface arrangements. Using the SISSO approach and DFT calculations, the adsorption energies of CO on the HEA AuCuIrPdPt(111) surfaces are predicted to identify improved catalysts for HCOOH decomposition, which provides a potential solution to hydrogen storage. We identify Au8Cu5IrPd21Pt13, Pd2Au2 and Pd2Au/Pd as promising candidates, and DFT calculations and microkinetic modeling show that the systems present superior activity to conventional Pd catalysts by three orders of magnitude at 400 K while maintaining high H2 selectivity. HCOOH decomposition proceeds through HCOO* and COOH* species on Au8Cu5IrPd21Pt13, Pd2Au2 and Pd2Au/Pd, with the rate-determining steps being HCOO* and COOH* dehydrogenation, respectively. Various PdAu surface alloys with varied Pd/Au ratios also exhibit salient activities, which agrees well with the superior performance of PdAu catalysts widely observed in experimental studies. This work highlights the importance of the ensemble effect in HCOOH decomposition, and the combination of a data-driven approach, DFT calculations and microkinetic modeling provides a powerful tool for fast catalyst discovery.
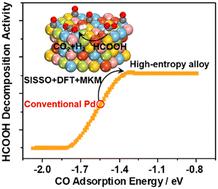
HCOOH分解高熵合金催化剂的机器学习辅助计算筛选
在许多催化反应中,催化活性与关键中间体的吸附能有关,基于这些中间体吸附能的计算筛选已被广泛用于催化剂的发现。高熵合金(HEAs)提供了广阔的构型空间,导致中间体的吸附能接近连续分布,从而有助于确定具有最佳吸附能的有前途的催化剂。然而,由于大量的表面排列,对HEAs吸附能的综合DFT计算受到阻碍。利用SISSO方法和DFT计算,预测了CO在HEA AuCuIrPdPt(111)表面的吸附能,以确定HCOOH分解的改进催化剂,从而为氢储存提供了潜在的解决方案。我们确定了Au8Cu5IrPd21Pt13, Pd2Au2和Pd2Au/Pd是有希望的候选者,DFT计算和微动力学建模表明,这些体系在400 K时具有比传统Pd催化剂优越的三个数量级的活性,同时保持了高的H2选择性。HCOOH的分解通过Au8Cu5IrPd21Pt13、Pd2Au2和Pd2Au/Pd上的HCOO*和COOH*物质进行,其速率决定步骤分别为HCOO*和COOH*脱氢。不同Pd/Au比的各种PdAu表面合金也表现出显著的活性,这与实验研究中广泛观察到的PdAu催化剂的优越性能相吻合。这项工作强调了系综效应在HCOOH分解中的重要性,并且将数据驱动方法、DFT计算和微动力学建模相结合,为快速发现催化剂提供了强大的工具。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Materials Chemistry A
CHEMISTRY, PHYSICAL-ENERGY & FUELS
CiteScore
19.50
自引率
5.00%
发文量
1892
审稿时长
1.5 months
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


