Seyda Gul Ozcan, Eylul Koc, Ahmet Murt, Mevlut Tamer Dincer, Iclal Gurses, Nurhan Seyahi, Sinan Trabulus
{"title":"A rare case of dual glomerular pathology: Alport syndrome and immune complex-mediated MPGN.","authors":"Seyda Gul Ozcan, Eylul Koc, Ahmet Murt, Mevlut Tamer Dincer, Iclal Gurses, Nurhan Seyahi, Sinan Trabulus","doi":"10.1186/s12882-025-04400-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN) and Alport syndrome are distinct glomerular diseases with different pathophysiologic mechanisms. Their coexistence is extremely rare and may present diagnostic and therapeutic challenges.</p><p><strong>Case presentation: </strong>A 42-year-old woman presented with persistent proteinuria and hematuria. Initial laboratory evaluations showed a progressive increase in proteinuria. She had a family history of renal disease. A renal biopsy, which had initially been postponed on patient request, subsequently revealed diffuse mesangial and focal endocapillary proliferation, GBM thickening, periglomerular fibrosis, and immune complex deposition with IgM, C3, and C1q, thereby confirming the diagnosis of IC-MPGN. Genetic testing identified a heterozygous COL4A5 c.1871G > A (p.Gly624Asp) mutation consistent with X-linked Alport syndrome. The patient follows conservative management now.</p><p><strong>Conclusion: </strong>This case highlights the rare coexistence of IC-MPGN, chronic TIN and Alport syndrome. In patients with progressive glomerular disease, especially with a family history of renal disease or extrarenal findings, genetic evaluation should be considered. Early renal biopsy and genetic testing are essential to guide management in complex cases with overlapping features.</p>","PeriodicalId":9089,"journal":{"name":"BMC Nephrology","volume":"26 1","pages":"514"},"PeriodicalIF":2.4000,"publicationDate":"2025-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12409941/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Nephrology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12882-025-04400-z","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN) and Alport syndrome are distinct glomerular diseases with different pathophysiologic mechanisms. Their coexistence is extremely rare and may present diagnostic and therapeutic challenges.
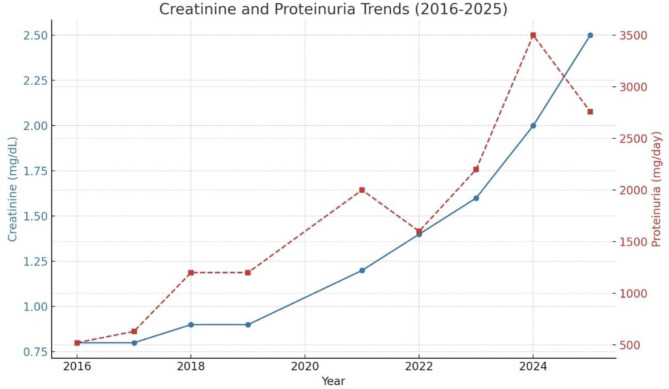
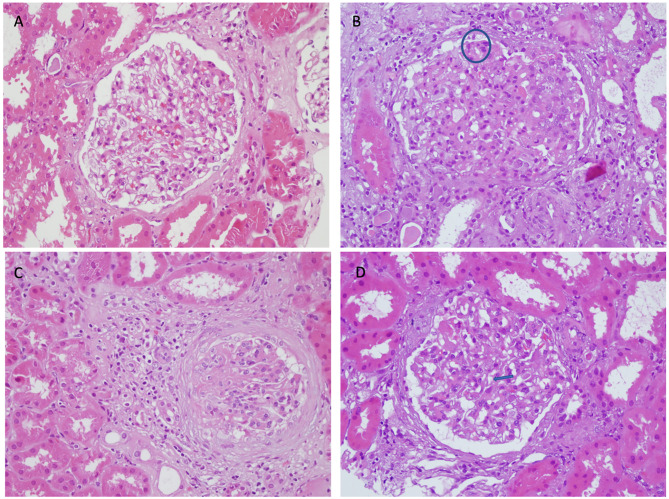
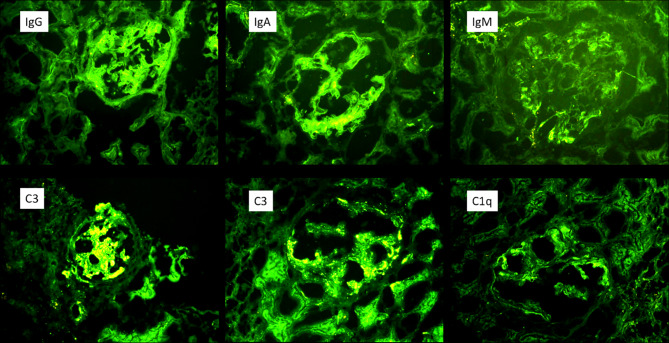
Case presentation: A 42-year-old woman presented with persistent proteinuria and hematuria. Initial laboratory evaluations showed a progressive increase in proteinuria. She had a family history of renal disease. A renal biopsy, which had initially been postponed on patient request, subsequently revealed diffuse mesangial and focal endocapillary proliferation, GBM thickening, periglomerular fibrosis, and immune complex deposition with IgM, C3, and C1q, thereby confirming the diagnosis of IC-MPGN. Genetic testing identified a heterozygous COL4A5 c.1871G > A (p.Gly624Asp) mutation consistent with X-linked Alport syndrome. The patient follows conservative management now.
Conclusion: This case highlights the rare coexistence of IC-MPGN, chronic TIN and Alport syndrome. In patients with progressive glomerular disease, especially with a family history of renal disease or extrarenal findings, genetic evaluation should be considered. Early renal biopsy and genetic testing are essential to guide management in complex cases with overlapping features.
期刊介绍:
BMC Nephrology is an open access journal publishing original peer-reviewed research articles in all aspects of the prevention, diagnosis and management of kidney and associated disorders, as well as related molecular genetics, pathophysiology, and epidemiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


