{"title":"Epidemiology and Genetic Diversity of Human Metapneumovirus in Patients with Severe Acute Respiratory Infection from 2023 to 2024 in Ningxia, China.","authors":"Ting Mu, Jianxin Pei, Jingting Wang, Ling Niu, Zhonglan Wu","doi":"10.3390/diseases13080255","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Human metapneumovirus (HMPV) is a major pathogen responsible for causing severe acute respiratory infections (SARI). Whole-genome sequencing can better identify transmission events and outbreaks. In this study, we aimed to investigate the epidemiology and genetic diversity of HMPV in SARI cases in Ningxia, China.</p><p><strong>Methods: </strong>We collected respiratory tract samples from hospitalized patients with SARI from October 2023 to September 2024 in Ningxia, China. Nasopharyngeal swabs were tested for respiratory viruses with qRT-PCR. Whole-genome sequences were determined for samples with high viral loads using an amplicon-based method.</p><p><strong>Results: </strong>We enrolled 2873 SARI patients from October 2023 to September 2024, and found an HMPV-positive proportion of 3.06% (88/2873). Children aged 4 years were particularly susceptible to HMPV infection, with a positive proportion of 10.92% (13/119). HMPV exhibits distinct seasonal characteristics, consistent with its established epidemiological pattern, with a peak incidence occurring during winter months. Sixteen complete HMPV genome sequences were obtained. Among these, 81.25% (13/16) were identified as genotype A (A2.2.2: 92.31%, 12/13; A2.2.1: 7.69%, 1/13) and 18.75% (3/16) as genotype B1. Notably, the dominant strain was 111nt-dup in genotype A2.2.2. Sequence analysis of HMPV genes revealed divergent G-gene sequence identities between different genotypes. Additionally, the potential glycosylation sites of the G protein varied across genotypes.</p><p><strong>Conclusions: </strong>In this study, we found that the 111nt-dup strain was the dominant one in genotype A, and multiple genotypes co-circulated in Ningxia from October 2023 to September 2024. The HMPV G protein exhibited the highest level of inter-strain diversity between genotypes. These findings provide valuable insights into the prevention and control of HMPV infections in China.</p>","PeriodicalId":72832,"journal":{"name":"Diseases (Basel, Switzerland)","volume":"13 8","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2025-08-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12385779/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Diseases (Basel, Switzerland)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/diseases13080255","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Human metapneumovirus (HMPV) is a major pathogen responsible for causing severe acute respiratory infections (SARI). Whole-genome sequencing can better identify transmission events and outbreaks. In this study, we aimed to investigate the epidemiology and genetic diversity of HMPV in SARI cases in Ningxia, China.
Methods: We collected respiratory tract samples from hospitalized patients with SARI from October 2023 to September 2024 in Ningxia, China. Nasopharyngeal swabs were tested for respiratory viruses with qRT-PCR. Whole-genome sequences were determined for samples with high viral loads using an amplicon-based method.
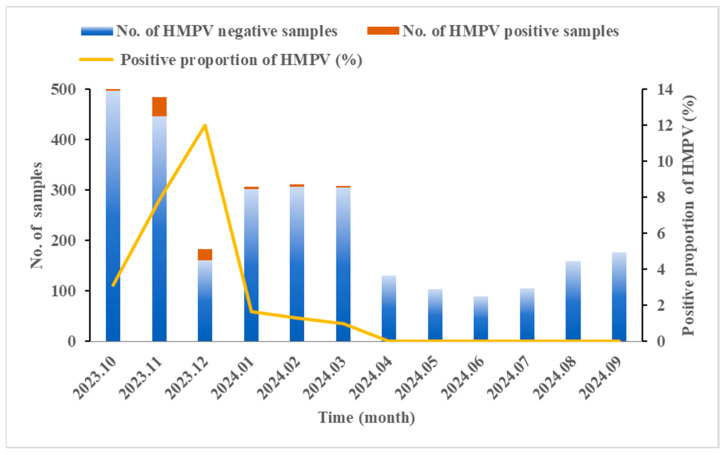
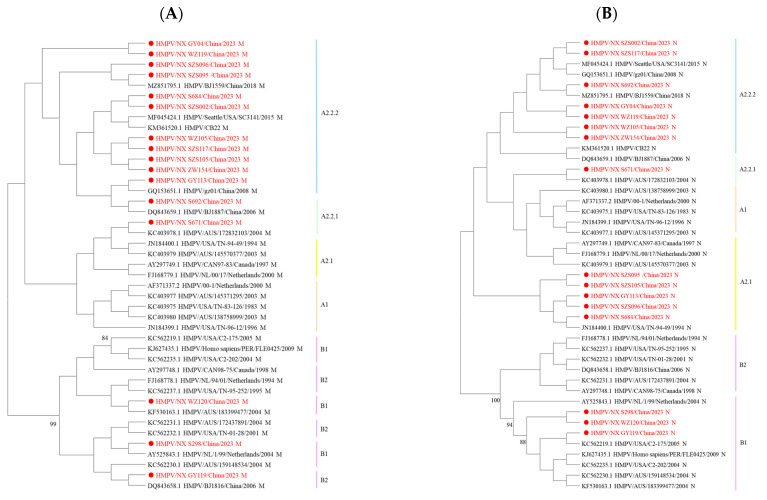
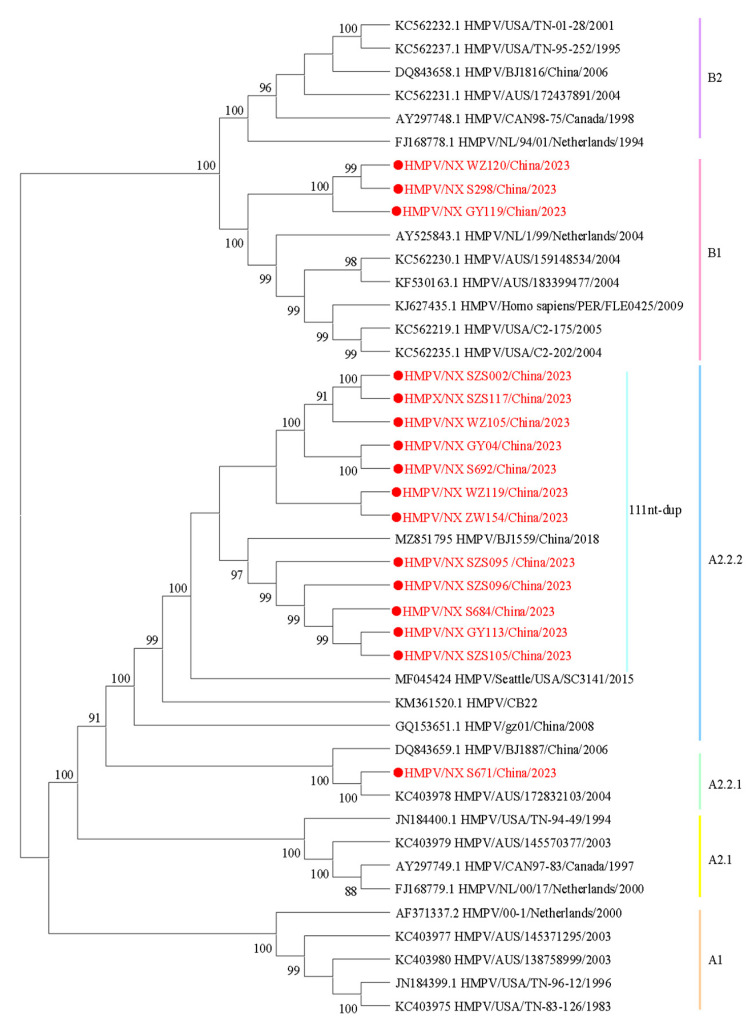
Results: We enrolled 2873 SARI patients from October 2023 to September 2024, and found an HMPV-positive proportion of 3.06% (88/2873). Children aged 4 years were particularly susceptible to HMPV infection, with a positive proportion of 10.92% (13/119). HMPV exhibits distinct seasonal characteristics, consistent with its established epidemiological pattern, with a peak incidence occurring during winter months. Sixteen complete HMPV genome sequences were obtained. Among these, 81.25% (13/16) were identified as genotype A (A2.2.2: 92.31%, 12/13; A2.2.1: 7.69%, 1/13) and 18.75% (3/16) as genotype B1. Notably, the dominant strain was 111nt-dup in genotype A2.2.2. Sequence analysis of HMPV genes revealed divergent G-gene sequence identities between different genotypes. Additionally, the potential glycosylation sites of the G protein varied across genotypes.
Conclusions: In this study, we found that the 111nt-dup strain was the dominant one in genotype A, and multiple genotypes co-circulated in Ningxia from October 2023 to September 2024. The HMPV G protein exhibited the highest level of inter-strain diversity between genotypes. These findings provide valuable insights into the prevention and control of HMPV infections in China.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


