Mayowa A Osundiji, Alicia Chen, Joseph D Farris, Radhika Dhamija
{"title":"Movement Disorder Following Hypoglycemic Encephalopathy in Mitochondrial 3-Hydroxy-3-methylglutaryl-CoA Synthase-2 (mHS) Deficiency.","authors":"Mayowa A Osundiji, Alicia Chen, Joseph D Farris, Radhika Dhamija","doi":"10.7326/aimcc.2025.0080","DOIUrl":null,"url":null,"abstract":"<p><p>Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase (mHS) deficiency is an ultra-rare inborn error of ketone body synthesis that is caused by biallelic mutations in <i>HMGCS2</i>. The manifestations of mHS deficiency can include hypoketotic hypoglycemia, metabolic acidosis, lethargy, encephalopathy, hyperammonemia, and hepatomegaly. Here, we report a case of movement disorder following hypoglycemic encephalopathy involving the basal ganglia in a patient with mHS deficiency. Exome sequencing showed novel compound heterozygous variants in <i>HMGCS2</i>, a partial gene deletion (classified as pathogenic) and c.704T>A (p.M235K) variant that was deemed to be likely pathogenic. Our findings suggest that mHS deficiency can result in basal ganglia injury and movement disorder.</p>","PeriodicalId":72222,"journal":{"name":"Annals of internal medicine. Clinical cases","volume":"4 8","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12377477/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of internal medicine. Clinical cases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.7326/aimcc.2025.0080","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/5 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
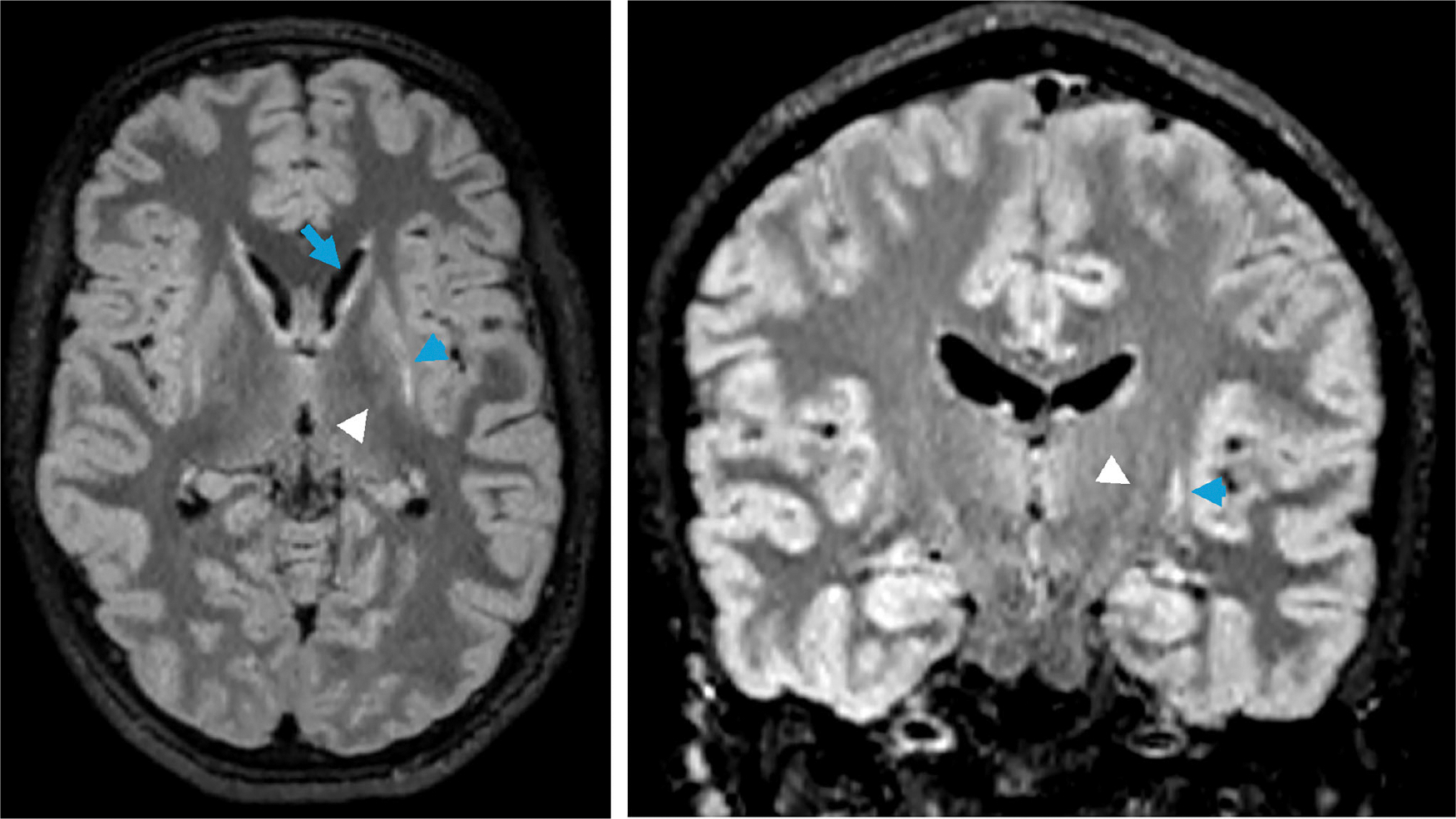
Mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase (mHS) deficiency is an ultra-rare inborn error of ketone body synthesis that is caused by biallelic mutations in HMGCS2. The manifestations of mHS deficiency can include hypoketotic hypoglycemia, metabolic acidosis, lethargy, encephalopathy, hyperammonemia, and hepatomegaly. Here, we report a case of movement disorder following hypoglycemic encephalopathy involving the basal ganglia in a patient with mHS deficiency. Exome sequencing showed novel compound heterozygous variants in HMGCS2, a partial gene deletion (classified as pathogenic) and c.704T>A (p.M235K) variant that was deemed to be likely pathogenic. Our findings suggest that mHS deficiency can result in basal ganglia injury and movement disorder.
线粒体3-羟基-3-甲基戊二酰辅酶A合成酶(mHS)缺乏症是一种极为罕见的先天性酮体合成错误,由HMGCS2的双等位基因突变引起。mHS缺乏的表现包括低酮性低血糖、代谢性酸中毒、嗜睡、脑病、高氨血症和肝肿大。在这里,我们报告一例低血糖性脑病累及基底神经节的mHS缺乏症患者的运动障碍。外显子组测序显示HMGCS2中存在新的复合杂合变异,部分基因缺失(归类为致病性)和c.704T> a (p.M235K)变异被认为可能具有致病性。我们的研究结果表明,mHS缺乏可导致基底神经节损伤和运动障碍。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


