Charlotte Mouraux, Tamara Dangouloff, Margaux Poleur, Laurane Mackels, Laura Vanden Brande, Aurore Daron, Laurent Servais, Alain Maertens de Noordhout, Stéphanie Delstanche
{"title":"Epidemiological report and diagnostic approach used in the neuromuscular population of Liege, Belgium.","authors":"Charlotte Mouraux, Tamara Dangouloff, Margaux Poleur, Laurane Mackels, Laura Vanden Brande, Aurore Daron, Laurent Servais, Alain Maertens de Noordhout, Stéphanie Delstanche","doi":"10.1186/s13023-025-03963-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Patients with neuromuscular diseases (NMD) have undergone considerable technological progress in terms of diagnosis and treatment over the past few years. Specifically, next-generation sequencing (NGS) has significantly expanded genetic diagnosis. Despite this, some patients remain undiagnosed and therefore without access to specific treatments. Analyses of epidemiology and diagnostic approaches in reference centers are required to determine effective strategies to improve diagnostic rates.</p><p><strong>Methods: </strong>We studied the proportion of each NMD and associated investigations in the patient population of the Neuromuscular Reference Center (NMRC) of Liege, Belgium, in 2023. The investigation tools used included laboratory testing, muscle biopsy, muscle imaging, single-gene sequencing, targeted NGS panels, and whole-exome sequencing (WES).</p><p><strong>Results: </strong>Of the 1084 patients who were regularly followed up, more than one-third had neuropathies (36.6%) that were divided equally between genetic and acquired causes. The second most common disorder was muscular dystrophies, which represented more than a quarter (27.5%). Third, 11.2% of the patients had motor neuron diseases. The other NMD (i.e., myopathies, ataxias, spastic paraplegias, and channelopathies) ranged from 2.1% to 6. %. A total of 13.7% of the patients had unconfirmed diagnoses, 31.5% had confirmed acquired disorders, and 54.9% had genetically confirmed disorders. Among the genetic diagnoses, 32.7% were obtained by NGS. The remaining 67.3% were determined using other genetic testing methods [i.e., array comparative genomic hybridization (aCGH), multiplex ligation-dependent probe amplification (MLPA), polymerase chain reaction (PCR), southern blotting (SB)].</p><p><strong>Conclusion: </strong>More than two-thirds of patients received a definitive diagnosis without the use of next-generation sequencing. Although innovative technologies such as whole genome sequencing and long-read sequencing are expected to eventually replace NGS panels and traditional methods (e.g., MLPA, PCR, aCGH), their current cost and the complexity of variant interpretation limit their widespread use in routine clinical practice. As a result, these older techniques remain relevant and valuable in current diagnostic workflow.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"464"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12398174/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03963-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Patients with neuromuscular diseases (NMD) have undergone considerable technological progress in terms of diagnosis and treatment over the past few years. Specifically, next-generation sequencing (NGS) has significantly expanded genetic diagnosis. Despite this, some patients remain undiagnosed and therefore without access to specific treatments. Analyses of epidemiology and diagnostic approaches in reference centers are required to determine effective strategies to improve diagnostic rates.
Methods: We studied the proportion of each NMD and associated investigations in the patient population of the Neuromuscular Reference Center (NMRC) of Liege, Belgium, in 2023. The investigation tools used included laboratory testing, muscle biopsy, muscle imaging, single-gene sequencing, targeted NGS panels, and whole-exome sequencing (WES).
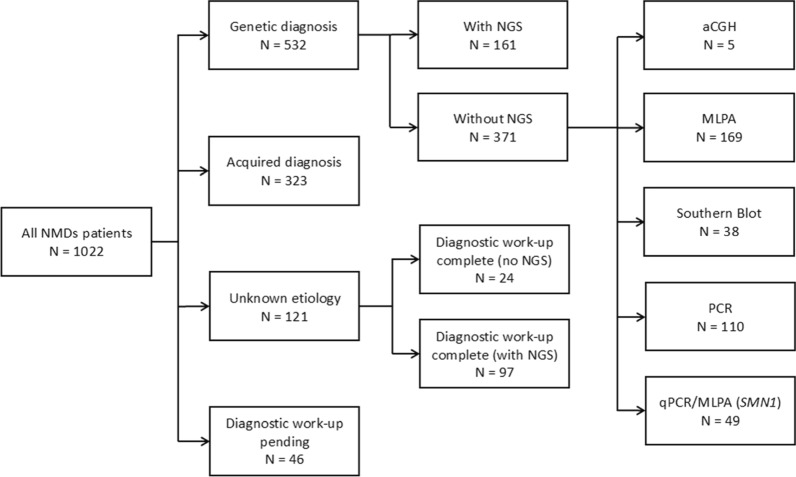
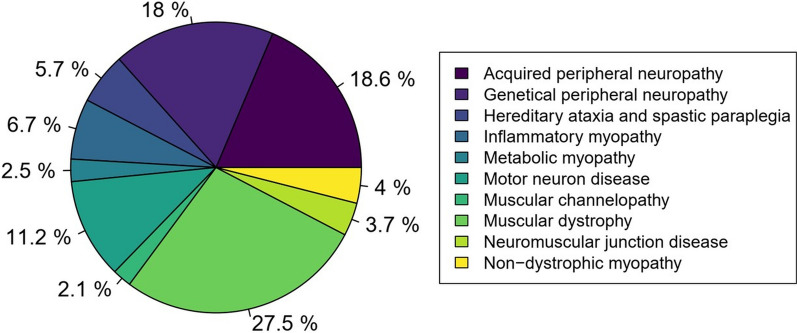
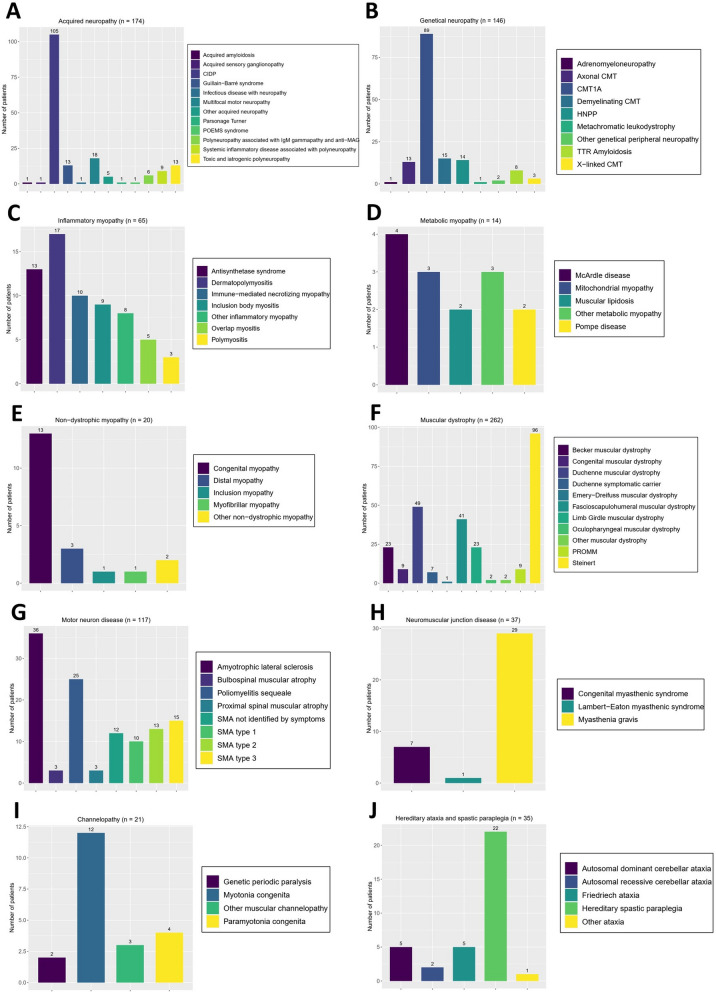
Results: Of the 1084 patients who were regularly followed up, more than one-third had neuropathies (36.6%) that were divided equally between genetic and acquired causes. The second most common disorder was muscular dystrophies, which represented more than a quarter (27.5%). Third, 11.2% of the patients had motor neuron diseases. The other NMD (i.e., myopathies, ataxias, spastic paraplegias, and channelopathies) ranged from 2.1% to 6. %. A total of 13.7% of the patients had unconfirmed diagnoses, 31.5% had confirmed acquired disorders, and 54.9% had genetically confirmed disorders. Among the genetic diagnoses, 32.7% were obtained by NGS. The remaining 67.3% were determined using other genetic testing methods [i.e., array comparative genomic hybridization (aCGH), multiplex ligation-dependent probe amplification (MLPA), polymerase chain reaction (PCR), southern blotting (SB)].
Conclusion: More than two-thirds of patients received a definitive diagnosis without the use of next-generation sequencing. Although innovative technologies such as whole genome sequencing and long-read sequencing are expected to eventually replace NGS panels and traditional methods (e.g., MLPA, PCR, aCGH), their current cost and the complexity of variant interpretation limit their widespread use in routine clinical practice. As a result, these older techniques remain relevant and valuable in current diagnostic workflow.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


