Giuseppe d'Orsi, Maria Teresa Di Claudio, Antonella Liantonio, Paola Imbrici, Cosimo Damiano Altomare, Orazio Palumbo, Pietro Palumbo, Mario Benvenuto, Nicola Gambacorta, Graziano Lolli, Massimo Carella
{"title":"Clinical course and management challenges in Lafora disease: a narrative analysis in an Apulian cohort.","authors":"Giuseppe d'Orsi, Maria Teresa Di Claudio, Antonella Liantonio, Paola Imbrici, Cosimo Damiano Altomare, Orazio Palumbo, Pietro Palumbo, Mario Benvenuto, Nicola Gambacorta, Graziano Lolli, Massimo Carella","doi":"10.1186/s13023-025-03976-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Lafora disease (LD) is an ultra-rare, autosomal recessive neurodegenerative disorder characterized by the accumulation of Lafora bodies in the brain, leading to drug-resistant epilepsy, myoclonus, progressive dementia, and cerebellar dysfunction. This retrospective study describes the clinical course and management challenges of LD in a cohort of patients from the Apulia region of Southern Italy, where the disease prevalence appears to be higher than in other populations.</p><p><strong>Methods: </strong>We retrospectively analyzed clinical, electroencephalographic, and management data from six unrelated families with a confirmed diagnosis of LD, followed at the Neurology Unit of the Scientific Institute Casa Sollievo della Sofferenza Hospital between 2010 and 2024. Demographic information, clinical presentation, treatment history, disease progression, and outcomes were collected.</p><p><strong>Results: </strong>Our analysis identified three distinct electroclinical stages: an initial Presenting Symptoms Stage with the onset of seizures and subsequent development of myoclonus; a Progressive Neurodegeneration Stage characterized by drug-resistant epilepsy, dementia, and ataxia; and a Terminal Stage marked by severe disability, frequent seizure emergencies, and medical complications. Management in the late stages proved particularly challenging, requiring a multidisciplinary approach to address refractory seizures, status epilepticus, and medical complications such as aspiration pneumonia and respiratory failure. Home-based care, with specialized team support, played a crucial role in minimizing hospitalizations.</p><p><strong>Discussion: </strong>Our findings underscore the importance of early diagnosis and a multidisciplinary approach in the management of LD. The late stages of the disease are characterized by significant clinical challenges necessitating close collaboration among neurologists, epileptologists, and other healthcare professionals, supported by effective home-based care. The apparent higher prevalence in Apulia warrants further investigation into potential genetic or environmental factors.</p><p><strong>Conclusion: </strong>This study highlights the significant clinical burden of LD and emphasizes the importance of multidisciplinary management, particularly in the advanced stages. Home-based care supported by specialized teams and caregivers is essential for optimizing patient well-being. Further research is needed to identify early biomarkers and develop targeted therapies for this devastating condition.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"447"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12369230/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03976-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Lafora disease (LD) is an ultra-rare, autosomal recessive neurodegenerative disorder characterized by the accumulation of Lafora bodies in the brain, leading to drug-resistant epilepsy, myoclonus, progressive dementia, and cerebellar dysfunction. This retrospective study describes the clinical course and management challenges of LD in a cohort of patients from the Apulia region of Southern Italy, where the disease prevalence appears to be higher than in other populations.
Methods: We retrospectively analyzed clinical, electroencephalographic, and management data from six unrelated families with a confirmed diagnosis of LD, followed at the Neurology Unit of the Scientific Institute Casa Sollievo della Sofferenza Hospital between 2010 and 2024. Demographic information, clinical presentation, treatment history, disease progression, and outcomes were collected.
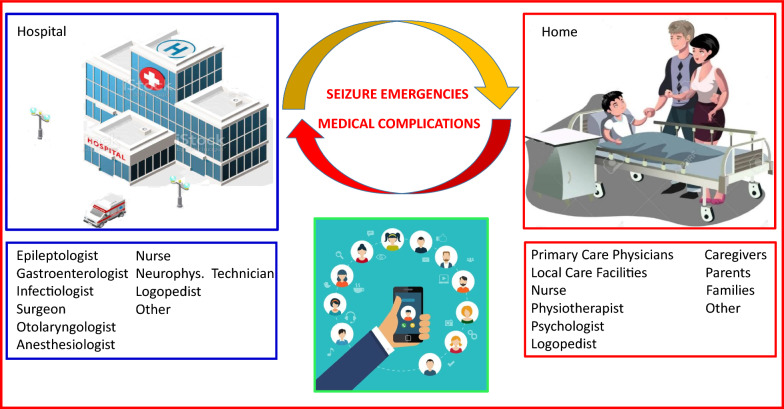
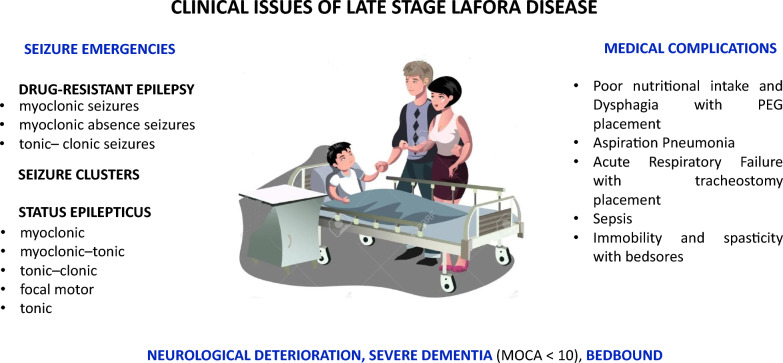
Results: Our analysis identified three distinct electroclinical stages: an initial Presenting Symptoms Stage with the onset of seizures and subsequent development of myoclonus; a Progressive Neurodegeneration Stage characterized by drug-resistant epilepsy, dementia, and ataxia; and a Terminal Stage marked by severe disability, frequent seizure emergencies, and medical complications. Management in the late stages proved particularly challenging, requiring a multidisciplinary approach to address refractory seizures, status epilepticus, and medical complications such as aspiration pneumonia and respiratory failure. Home-based care, with specialized team support, played a crucial role in minimizing hospitalizations.
Discussion: Our findings underscore the importance of early diagnosis and a multidisciplinary approach in the management of LD. The late stages of the disease are characterized by significant clinical challenges necessitating close collaboration among neurologists, epileptologists, and other healthcare professionals, supported by effective home-based care. The apparent higher prevalence in Apulia warrants further investigation into potential genetic or environmental factors.
Conclusion: This study highlights the significant clinical burden of LD and emphasizes the importance of multidisciplinary management, particularly in the advanced stages. Home-based care supported by specialized teams and caregivers is essential for optimizing patient well-being. Further research is needed to identify early biomarkers and develop targeted therapies for this devastating condition.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


