{"title":"Prevalence and predictors of uncommon features in FSHD1 patients: insights from the French FSHD registry.","authors":"Benoît Sanson, Abderhmane Slioui, Jérémy Garcia, Lori Klouvi, Julie Lejeune, Caroline Stalens, Céline Guien, Sitraka Rabarimeriarijaona, Rafaëlle Bernard, Juliette Nectoux, Sharham Attarian, Anne-Laure Bédat-Millet, Françoise Bouhour, François Constant Boyer, Jean-Baptiste Chanson, Ariane Choumert, Pascal Cintas, Elisa De La Cruz, Léonard Féasson, Maxime Fournier, Karima Ghorab, Agnès Jacquin-Piques, Pascal Laforêt, Armelle Magot, Maud Michaud, Jean-Baptiste Noury, Guilhem Solé, Marco Spinazzi, Tanya Stojkovic, Céline Tard, Luisa Villa, Christophe Béroud, Sabrina Sacconi","doi":"10.1186/s13023-025-03877-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Facioscapulohumeral muscular dystrophy (FSHD) is characterized by a typical pattern of muscle involvement, yet it encompasses a wide spectrum of phenotypes, including less common features that remain incompletely defined in the literature. While previous studies have highlighted this clinical variability, no consensus has been reached on how to classify uncommon manifestations, nor have specific predictors been identified. This study aims to describe these uncommon features and explore potential predictors, utilizing data from the French FSHD registry. To this end, we analysed data from 306 FSHD1 patients across nine French neuromuscular referral centres. Descriptive statistics, univariate analyses, and multiple logistic regression models were employed to examine uncommon characteristics and their predictors.</p><p><strong>Results: </strong>Uncommon features were observed in 19.6% of cases. The most common was a discrepancy between disease severity and D4Z4 repeat unit (RU) count (41.7%), followed by predominant impairment at proximal lower limb or distal upper limb muscles (21.7%). Three unanticipated features emerged: isolated or predominant axial impairment, anosmia and atopic dermatitis. Univariate analysis revealed that uncommon features were associated with higher RU count (6.5 ± 2.1 vs. 5.8 ± 1.8 in typical patients) and older age of onset (32.0 ± 18.8 years vs. 25.0 ± 15.4 years). Such features were more prevalent in the borderline 8-10 RU range, an association confirmed by multivariate analysis (OR = 2.43, 95% CI 1.21 to 4.87). Later age of onset consistently emerged as a factor across multiple multivariate models.</p><p><strong>Conclusions: </strong>This study documents uncommon FSHD features, revealing their association with the 8-10 RU range and later age of onset. These findings further support a complex interplay among genetic and epigenetic modifiers and ageing in shaping the clinical phenotype of FSHD, especially in patients carrying borderline D4Z4 arrays. Differential phenotypes, particularly in relation to RU range and age of onset, points to the importance of harmonized, comprehensive clinical and genetic assessments. Recognizing uncommon features may improve diagnostic accuracy and guide individualized management strategies, highlighting the need for tailored approaches to patient care.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"470"},"PeriodicalIF":3.5000,"publicationDate":"2025-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12403360/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03877-z","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Facioscapulohumeral muscular dystrophy (FSHD) is characterized by a typical pattern of muscle involvement, yet it encompasses a wide spectrum of phenotypes, including less common features that remain incompletely defined in the literature. While previous studies have highlighted this clinical variability, no consensus has been reached on how to classify uncommon manifestations, nor have specific predictors been identified. This study aims to describe these uncommon features and explore potential predictors, utilizing data from the French FSHD registry. To this end, we analysed data from 306 FSHD1 patients across nine French neuromuscular referral centres. Descriptive statistics, univariate analyses, and multiple logistic regression models were employed to examine uncommon characteristics and their predictors.
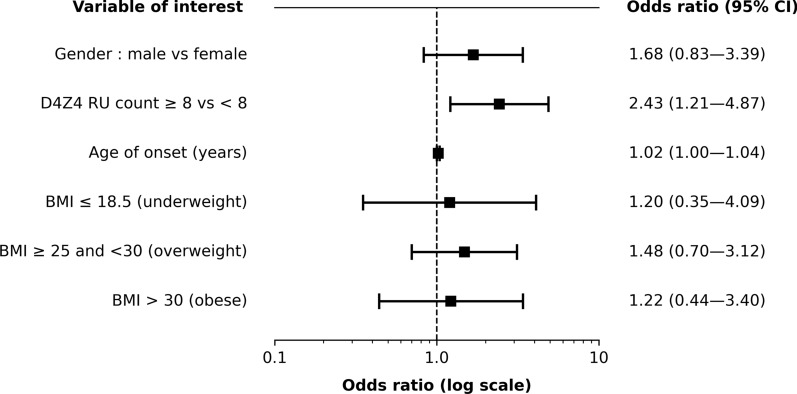
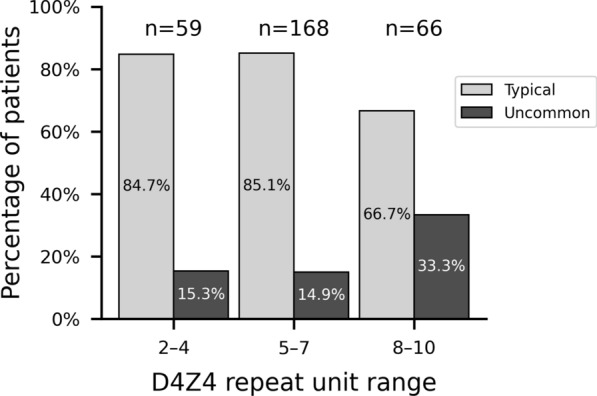
Results: Uncommon features were observed in 19.6% of cases. The most common was a discrepancy between disease severity and D4Z4 repeat unit (RU) count (41.7%), followed by predominant impairment at proximal lower limb or distal upper limb muscles (21.7%). Three unanticipated features emerged: isolated or predominant axial impairment, anosmia and atopic dermatitis. Univariate analysis revealed that uncommon features were associated with higher RU count (6.5 ± 2.1 vs. 5.8 ± 1.8 in typical patients) and older age of onset (32.0 ± 18.8 years vs. 25.0 ± 15.4 years). Such features were more prevalent in the borderline 8-10 RU range, an association confirmed by multivariate analysis (OR = 2.43, 95% CI 1.21 to 4.87). Later age of onset consistently emerged as a factor across multiple multivariate models.
Conclusions: This study documents uncommon FSHD features, revealing their association with the 8-10 RU range and later age of onset. These findings further support a complex interplay among genetic and epigenetic modifiers and ageing in shaping the clinical phenotype of FSHD, especially in patients carrying borderline D4Z4 arrays. Differential phenotypes, particularly in relation to RU range and age of onset, points to the importance of harmonized, comprehensive clinical and genetic assessments. Recognizing uncommon features may improve diagnostic accuracy and guide individualized management strategies, highlighting the need for tailored approaches to patient care.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


