Silvia Boeri, Maria Piai, Silvia Russo, Valentina Alari, Francesca Cogliati, Davide Simonetta, Timothy A Benke, Lino Nobili, Giulia Prato
{"title":"Clinical differences in monozygotic twins with Rett syndrome: case report and systematic review.","authors":"Silvia Boeri, Maria Piai, Silvia Russo, Valentina Alari, Francesca Cogliati, Davide Simonetta, Timothy A Benke, Lino Nobili, Giulia Prato","doi":"10.1186/s13023-025-03935-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Rett Syndrome (RTT) is a rare, and severe neurodevelopmental disorder that primarily affects females and is primarily (> 96%) due to pathogenic loss-of-function genetic variants of methyl-CpG-binding protein 2 (MECP2). Despite the rarity of the syndrome, sporadic twin cases have been reported. The descriptions have often focused on the phenotype, emphasizing differences or similarities. We report the case of monozygotic (MZ) twins with RTT carrying the same MECP2 mutation and perform a systematic review of the cases of MZ twins.</p><p><strong>Method: </strong>We searched PubMed and Embase for articles reporting MZ twins with RTT who met Neul criteria and carried mutations in the MECP2 gene. We focused on phenotypic discordance and X chromosome inactivation (XCI).</p><p><strong>Results: </strong>Our search yielded 115 results, 18 of which were included in our systematic review. We identified 17 pairs of twins, with 11 showing a discordant phenotype. Data on XCI were reported for only six pairs. We describe MZ twins with typical RTT syndrome who shared the same p.Thr158Met pathogenic variant on the MECP2 gene but exhibited different severity of clinical phenotype, especially regarding epilepsy. The XCI pattern and expression of the wild-type allele in blood were similar in both twins, suggesting that XCI differences assessed in blood may not account for the phenotypic variability. Mononucleate cells were isolated from both twins to generate induced pluripotent stem cells (iPSCs). The patient with more mutated clones presented a more severe phenotype.</p><p><strong>Discussion: </strong>Cases of MZ twins with RTT are few, and the phenotypic difference described in our case and presented in the literature does not seem to be explained by different XCI patterns. Therefore, more detailed genetic investigations are necessary.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"473"},"PeriodicalIF":3.5000,"publicationDate":"2025-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12406456/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03935-6","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Rett Syndrome (RTT) is a rare, and severe neurodevelopmental disorder that primarily affects females and is primarily (> 96%) due to pathogenic loss-of-function genetic variants of methyl-CpG-binding protein 2 (MECP2). Despite the rarity of the syndrome, sporadic twin cases have been reported. The descriptions have often focused on the phenotype, emphasizing differences or similarities. We report the case of monozygotic (MZ) twins with RTT carrying the same MECP2 mutation and perform a systematic review of the cases of MZ twins.
Method: We searched PubMed and Embase for articles reporting MZ twins with RTT who met Neul criteria and carried mutations in the MECP2 gene. We focused on phenotypic discordance and X chromosome inactivation (XCI).
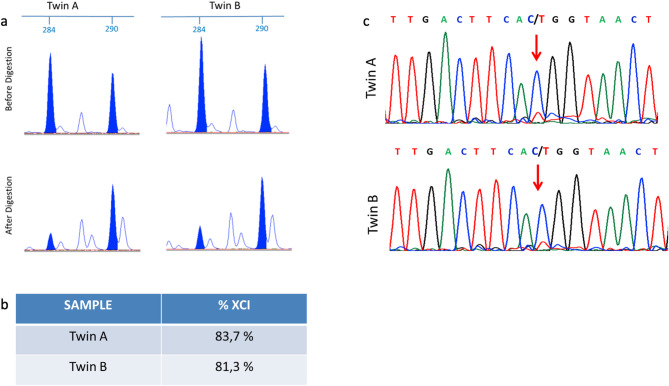
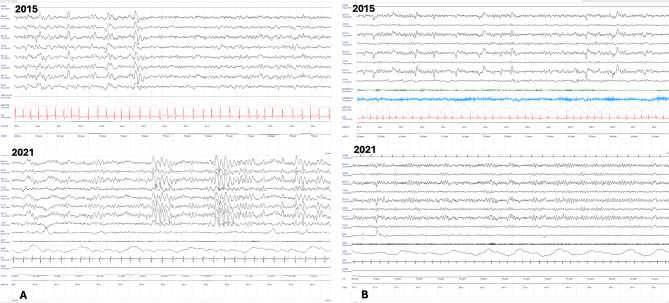
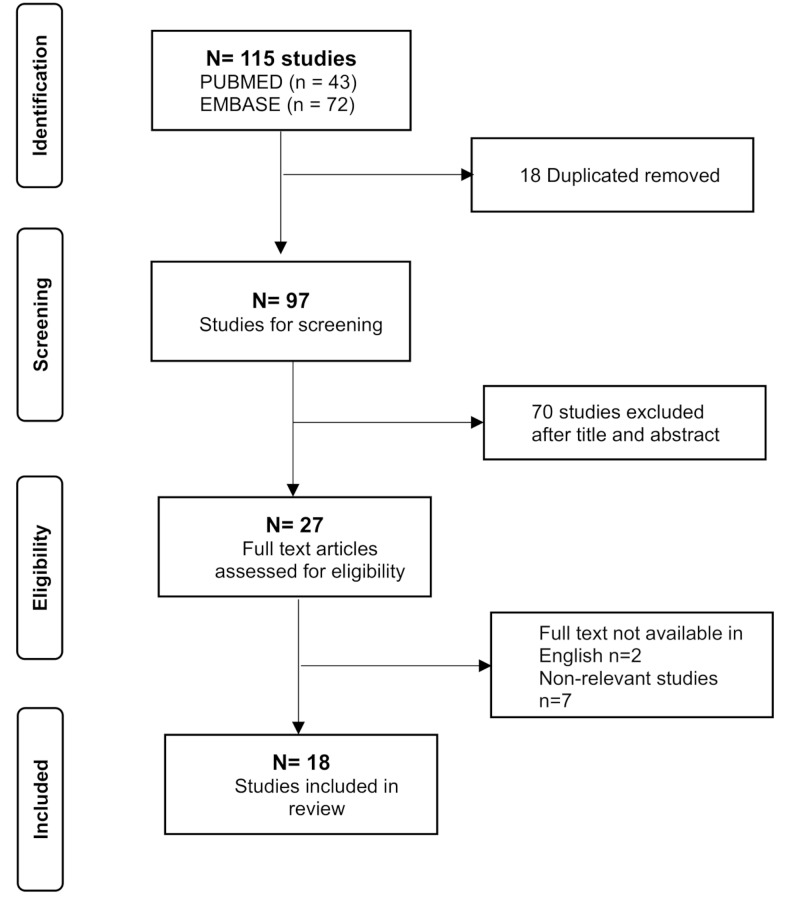
Results: Our search yielded 115 results, 18 of which were included in our systematic review. We identified 17 pairs of twins, with 11 showing a discordant phenotype. Data on XCI were reported for only six pairs. We describe MZ twins with typical RTT syndrome who shared the same p.Thr158Met pathogenic variant on the MECP2 gene but exhibited different severity of clinical phenotype, especially regarding epilepsy. The XCI pattern and expression of the wild-type allele in blood were similar in both twins, suggesting that XCI differences assessed in blood may not account for the phenotypic variability. Mononucleate cells were isolated from both twins to generate induced pluripotent stem cells (iPSCs). The patient with more mutated clones presented a more severe phenotype.
Discussion: Cases of MZ twins with RTT are few, and the phenotypic difference described in our case and presented in the literature does not seem to be explained by different XCI patterns. Therefore, more detailed genetic investigations are necessary.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


