Multiple overlapping binding sites determine transcription factor occupancy
IF 48.5
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
Abstract
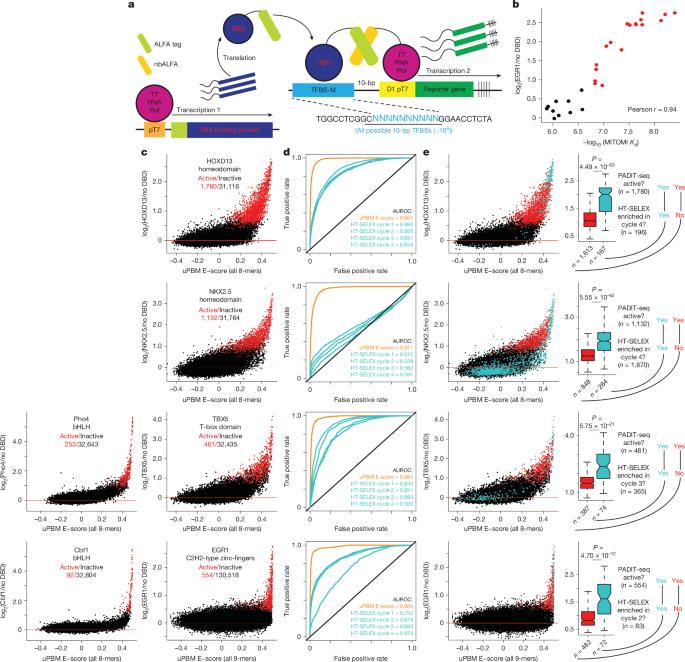
Transcription factors (TFs) regulate gene expression by interacting with DNA in a sequence-specific manner. High-throughput in vitro technologies, such as protein-binding microarrays1–6 and HT-SELEX (high-throughput systematic evolution of ligands by exponential enrichment)7,8, have revealed the DNA-binding specificities of hundreds of TFs. However, they have limited ability to reliably identify lower-affinity DNA binding sites, which are increasingly recognized as important for precise spatiotemporal control of gene expression9–19. Here, to address this limitation, we developed protein affinity to DNA by in vitro transcription and RNA sequencing (PADIT-seq), with which we comprehensively assayed the binding preferences of six TFs to all possible ten-base-pair DNA sequences, detecting hundreds of novel, lower-affinity binding sites. The expanded repertoire of lower-affinity binding sites revealed that nucleotides flanking high-affinity DNA binding sites create overlapping lower-affinity sites that together modulate TF genomic occupancy in vivo. We propose a model in which TF binding is not determined by individual binding sites, but rather by the sum of multiple, overlapping binding sites. The overlapping binding model explains how competition between paralogous TFs for shared high-affinity binding sites is determined by flanking nucleotides that create differential numbers of overlapping, lower-affinity binding sites. Critically, the model transforms our understanding of noncoding-variant effects, revealing how single nucleotide changes simultaneously alter multiple overlapping sites to additively influence gene expression and human traits, including diseases. A new method enables comprehensive screening and identification of low-affinity DNA binding sites for transcription factors, and reveals that nucleotides flanking high-affinity binding sites create overlapping low-affinity binding sites that modulate transcription factor binding in vivo.
多个重叠的结合位点决定了转录因子的占用。
转录因子(TFs)通过与DNA以序列特异性的方式相互作用来调节基因表达。高通量体外技术,如蛋白质结合微阵列技术1-6和HT-SELEX(高通量配体系统进化指数富集)7,8,已经揭示了数百种tf的dna结合特异性。然而,它们可靠地识别低亲和力DNA结合位点的能力有限,而低亲和力DNA结合位点对于基因表达的精确时空控制越来越重要-19。在这里,为了解决这一限制,我们通过体外转录和RNA测序(PADIT-seq)开发了蛋白质对DNA的亲和力,我们全面分析了6个tf对所有可能的10碱基对DNA序列的结合偏好,检测了数百个新的低亲和力结合位点。低亲和力结合位点的扩展库显示,高亲和力DNA结合位点两侧的核苷酸产生重叠的低亲和力位点,共同调节TF基因组在体内的占用。我们提出了一个模型,其中TF结合不是由单个结合位点决定的,而是由多个重叠结合位点的总和决定的。重叠结合模型解释了同源tf之间对共享高亲和力结合位点的竞争是如何由侧翼核苷酸决定的,这些核苷酸会产生不同数量的重叠低亲和力结合位点。关键的是,该模型改变了我们对非编码变异效应的理解,揭示了单个核苷酸变化如何同时改变多个重叠位点,进而影响基因表达和人类特征,包括疾病。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature
综合性期刊-综合性期刊
CiteScore
90.00
自引率
1.20%
发文量
3652
审稿时长
3 months
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


