Sonam Dukda, Manoharan Kumar, Andrew Calcino, Ulf Schmitz, Matt A Field
{"title":"Increasing pathogenic germline variant diagnosis rates in precision medicine: current best practices and future opportunities.","authors":"Sonam Dukda, Manoharan Kumar, Andrew Calcino, Ulf Schmitz, Matt A Field","doi":"10.1186/s40246-025-00811-z","DOIUrl":null,"url":null,"abstract":"<p><p>The accurate diagnosis of pathogenic variants is essential for effective clinical decision making within precision medicine programs. Despite significant advances in both the quality and quantity of molecular patient data, diagnostic rates remain suboptimal for many inherited diseases. As such, prioritisation and identification of pathogenic disease-causing variants remains a complex and rapidly evolving field. This review explores the latest technological and computational options being used to increase genetic diagnosis rates in precision medicine programs.While interpreting genetic variation via standards such as ACMG guidelines is increasingly being recognized as a gold standard approach, the underlying datasets and algorithms recommended are often slow to incorporate additional data types and methodologies. For example, new technological developments, particularly in single-cell and long-read sequencing, offer great opportunity to improve genetic diagnosis rates, however, how to best interpret and integrate increasingly complex multi-omics patient data remains unclear. Further, advances in artificial intelligence and machine learning applications in biomedical research offer enormous potential, however they require careful consideration and benchmarking given the clinical nature of the data. This review covers the current state of the art in available sequencing technologies, software methodologies for variant annotation/prioritisation, pedigree-based strategies and the potential role of machine learning applications. We describe a key set of design principles required for a modern multi-omic precision medicine framework that is robust, modular, secure, flexible, and scalable. Creating a next generation framework will ensure we realise the full potential of precision medicine into the future.</p>","PeriodicalId":13183,"journal":{"name":"Human Genomics","volume":"19 1","pages":"97"},"PeriodicalIF":4.3000,"publicationDate":"2025-08-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12374290/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40246-025-00811-z","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
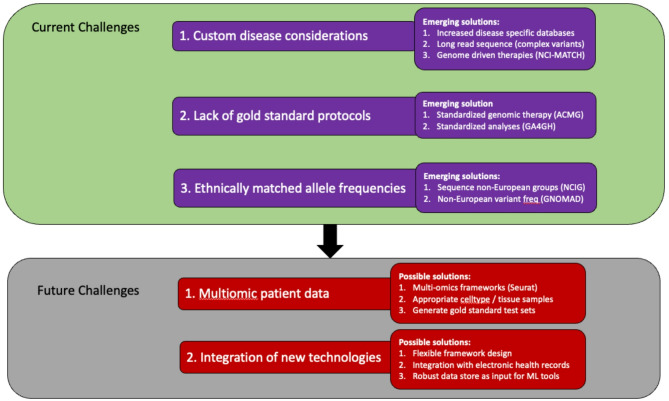
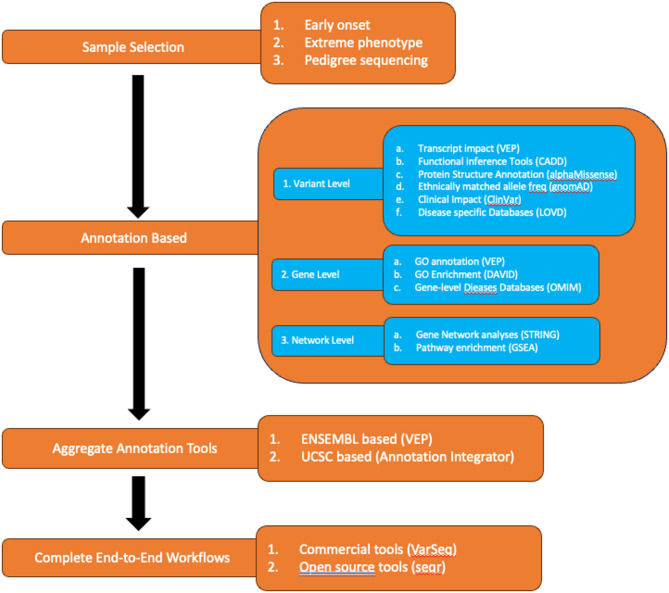
The accurate diagnosis of pathogenic variants is essential for effective clinical decision making within precision medicine programs. Despite significant advances in both the quality and quantity of molecular patient data, diagnostic rates remain suboptimal for many inherited diseases. As such, prioritisation and identification of pathogenic disease-causing variants remains a complex and rapidly evolving field. This review explores the latest technological and computational options being used to increase genetic diagnosis rates in precision medicine programs.While interpreting genetic variation via standards such as ACMG guidelines is increasingly being recognized as a gold standard approach, the underlying datasets and algorithms recommended are often slow to incorporate additional data types and methodologies. For example, new technological developments, particularly in single-cell and long-read sequencing, offer great opportunity to improve genetic diagnosis rates, however, how to best interpret and integrate increasingly complex multi-omics patient data remains unclear. Further, advances in artificial intelligence and machine learning applications in biomedical research offer enormous potential, however they require careful consideration and benchmarking given the clinical nature of the data. This review covers the current state of the art in available sequencing technologies, software methodologies for variant annotation/prioritisation, pedigree-based strategies and the potential role of machine learning applications. We describe a key set of design principles required for a modern multi-omic precision medicine framework that is robust, modular, secure, flexible, and scalable. Creating a next generation framework will ensure we realise the full potential of precision medicine into the future.
期刊介绍:
Human Genomics is a peer-reviewed, open access, online journal that focuses on the application of genomic analysis in all aspects of human health and disease, as well as genomic analysis of drug efficacy and safety, and comparative genomics.
Topics covered by the journal include, but are not limited to: pharmacogenomics, genome-wide association studies, genome-wide sequencing, exome sequencing, next-generation deep-sequencing, functional genomics, epigenomics, translational genomics, expression profiling, proteomics, bioinformatics, animal models, statistical genetics, genetic epidemiology, human population genetics and comparative genomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


