Comparing survival outcomes of anti-fibrotic therapy for idiopathic pulmonary fibrosis with and without emphysema: a multi-center real-world study from Taiwan.
{"title":"Comparing survival outcomes of anti-fibrotic therapy for idiopathic pulmonary fibrosis with and without emphysema: a multi-center real-world study from Taiwan.","authors":"Yu-Hung Fang, Yi-An Hsieh, Yen-Fu Chen, Yu-Chi Chiu, Yu-Ching Lin, Kuo-Tung Huang, Yung-Chia Huang, Yu-Feng Wei, Chien-Wen Huang, Pin-Kuei Fu","doi":"10.1186/s12890-025-03890-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Whether the long-term survival of patients with idiopathic pulmonary fibrosis (IPF) is worse than that of patients with IPF combined with emphysema after anti-fibrotic therapy is unclear. This study aimed to compare treatment outcomes between the two groups and identify potential predictors of mortality.</p><p><strong>Methods: </strong>This retrospective cohort study was conducted in seven hospitals across Taiwan between August 2015 and August 2022 and included patients with IPF who received anti-fibrotic agents covered by national insurance. Based on the extent of emphysema observed on high-resolution chest tomography, patients with IPF were categorized into two groups: IPF only; and IPF with emphysema. Baseline characteristics and survival outcomes were compared between the groups. Cox proportional hazards models were used for multivariable analysis to identify factors associated with overall mortality during the follow-up period.</p><p><strong>Results: </strong>Of the 275 patients included, 126 (45.8%) had IPF with emphysema and 149 (54.2%) had IPF only. The emphysema group had a higher proportion of males and patients with a smoking history, finger clubbing, comorbidities, or a definite usual interstitial pneumonia (UIP) pattern compared to the IPF-only group. Additionally, this group had a higher forced vital capacity (FVC, %) and forced expiratory volume in 1 s (FEV₁, L), while FEV₁ (%) was similar and FEV₁/FVC (%) was lower. During a median follow-up of 3.7 years, the overall survival rates were comparable (IPF only: 45.6%; IPF with emphysema: 48.4%). The overall survival of patients with probable UIP was significantly better than that of patients with definite UIP (53.5% vs. 34.6%). Likewise, the survival rate of the group with a diffusing capacity of the lung for carbon monoxide (DLCO) > 49% was higher than that of the group with DLCO ≤ 49% (53.9% vs. 31.4%). After adjusting for confounders, lower body mass index (BMI) (adjusted hazard ratio [aHR] = 0.95) and comorbid pulmonary hypertension (aHR = 2.27) were independently associated with increased overall mortality. Neither the presence of emphysema nor the type of anti-fibrotic agent was associated with mortality.</p><p><strong>Conclusions: </strong>The survival outcomes of patients with IPF and emphysema and those of patients with IPF only are comparable after treatment with anti-fibrotic agents. Lower BMI and comorbid pulmonary hypertension are significant predictors of increased mortality.</p>","PeriodicalId":9148,"journal":{"name":"BMC Pulmonary Medicine","volume":"25 1","pages":"401"},"PeriodicalIF":2.8000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12369227/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Pulmonary Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12890-025-03890-9","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"RESPIRATORY SYSTEM","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Whether the long-term survival of patients with idiopathic pulmonary fibrosis (IPF) is worse than that of patients with IPF combined with emphysema after anti-fibrotic therapy is unclear. This study aimed to compare treatment outcomes between the two groups and identify potential predictors of mortality.
Methods: This retrospective cohort study was conducted in seven hospitals across Taiwan between August 2015 and August 2022 and included patients with IPF who received anti-fibrotic agents covered by national insurance. Based on the extent of emphysema observed on high-resolution chest tomography, patients with IPF were categorized into two groups: IPF only; and IPF with emphysema. Baseline characteristics and survival outcomes were compared between the groups. Cox proportional hazards models were used for multivariable analysis to identify factors associated with overall mortality during the follow-up period.
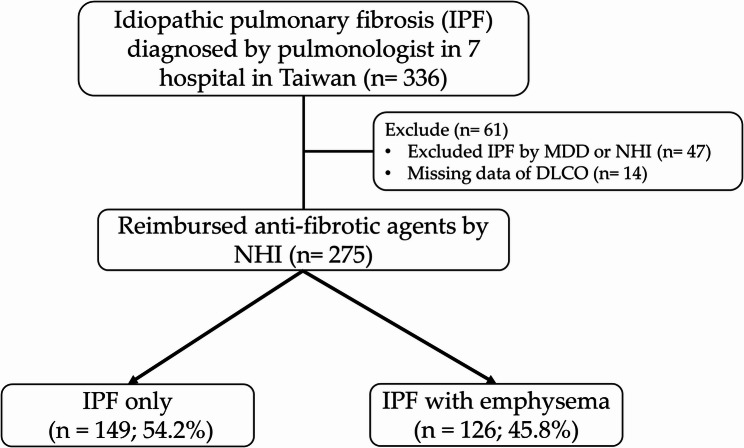
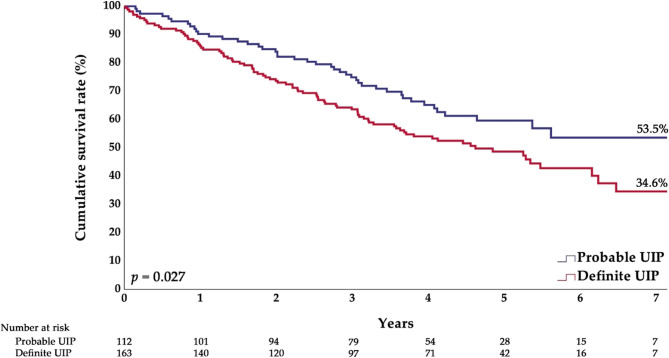
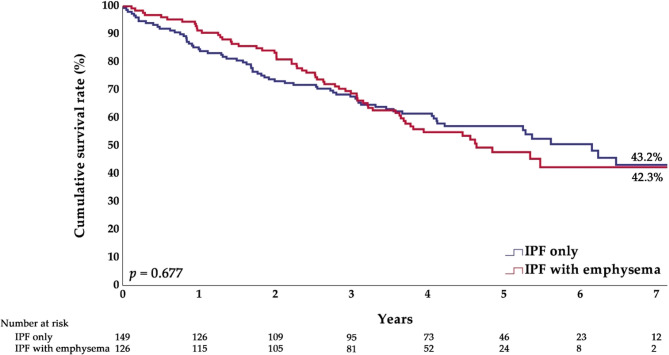
Results: Of the 275 patients included, 126 (45.8%) had IPF with emphysema and 149 (54.2%) had IPF only. The emphysema group had a higher proportion of males and patients with a smoking history, finger clubbing, comorbidities, or a definite usual interstitial pneumonia (UIP) pattern compared to the IPF-only group. Additionally, this group had a higher forced vital capacity (FVC, %) and forced expiratory volume in 1 s (FEV₁, L), while FEV₁ (%) was similar and FEV₁/FVC (%) was lower. During a median follow-up of 3.7 years, the overall survival rates were comparable (IPF only: 45.6%; IPF with emphysema: 48.4%). The overall survival of patients with probable UIP was significantly better than that of patients with definite UIP (53.5% vs. 34.6%). Likewise, the survival rate of the group with a diffusing capacity of the lung for carbon monoxide (DLCO) > 49% was higher than that of the group with DLCO ≤ 49% (53.9% vs. 31.4%). After adjusting for confounders, lower body mass index (BMI) (adjusted hazard ratio [aHR] = 0.95) and comorbid pulmonary hypertension (aHR = 2.27) were independently associated with increased overall mortality. Neither the presence of emphysema nor the type of anti-fibrotic agent was associated with mortality.
Conclusions: The survival outcomes of patients with IPF and emphysema and those of patients with IPF only are comparable after treatment with anti-fibrotic agents. Lower BMI and comorbid pulmonary hypertension are significant predictors of increased mortality.
期刊介绍:
BMC Pulmonary Medicine is an open access, peer-reviewed journal that considers articles on all aspects of the prevention, diagnosis and management of pulmonary and associated disorders, as well as related molecular genetics, pathophysiology, and epidemiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


