{"title":"Evaluation of the HumanMethylationEPIC v2.0 Bead Chip Using Low Quality and Quantity DNA Samples.","authors":"Brando Poggiali, Mikkel Eriksen Dupont, Marie-Louise Kampmann, Athina Vidaki, Vania Pereira, Claus Børsting, Jacob Tfelt-Hansen, Jeppe Dyrberg Andersen","doi":"10.1186/s12575-025-00292-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The HumanMethylationEPIC v2.0 BeadChip (EPIC v2.0) microarray is a widely used tool for genome-wide DNA methylation (DNAm) analysis, designed for high-quality human DNA with a recommended input of 250 ng. However, in clinical and forensic settings, DNA samples may be of low quality and/or quantity (highly fragmented and/or available in low amounts). This study assessed the performance of the EPIC v2.0 on DNA samples with various combinations of average DNA fragment size (350, 230, 165, and 95 bp) and DNA input amount (100, 50, 20, and 10 ng), compared to a paired control sample analyzed under optimal conditions (high-quality DNA and 250 ng DNA input).</p><p><strong>Results: </strong>The best performance was obtained for samples with average DNA fragment size of 350 bp and 100 ng DNA input (~ 90% probe detection rate, r = 0.995, and median absolute beta value differences|Δβ| = 0.012 when compared with the control sample). Samples with lower average DNA fragment sizes and DNA input amount performed worse, with the lowest probe detection rate (~ 43%), r = 0.946, and the highest|Δβ| (0.038). Samples with average DNA fragment sizes of 95 bp and those with 165 bp at 10 ng DNA input failed to pass sample quality control (QC). CpG sites with intermediate DNAm values (β = 0.1-0.9) showed higher|Δβ| than the extreme DNAm values (β = 0-0.1, and β = 0.9-1). Finally, we assessed an application of DNAm by performing epigenetic age analysis, and observed mean absolute errors (MAEs) below 10 years for 350 bp samples across four epigenetic clocks.</p><p><strong>Conclusions: </strong>Both DNA fragment size and DNA input amounts affect DNAm analysis on the EPIC v2.0, with the investigated DNA fragment size having a greater impact than the investigated DNA input amount. DNAm measurements were achieved with the EPIC v2.0 microarray down to an average DNA fragment size of 165 bp and a 20 ng DNA input. Highly fragmented DNA (95 bp) did not result in usable DNAm analysis as all samples failed QC. Overall, our study demonstrates the potential and limitations of EPIC v2.0 microarray with low quality and quantity DNA samples.</p>","PeriodicalId":8960,"journal":{"name":"Biological Procedures Online","volume":"27 1","pages":"34"},"PeriodicalIF":4.3000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12369268/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biological Procedures Online","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12575-025-00292-3","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
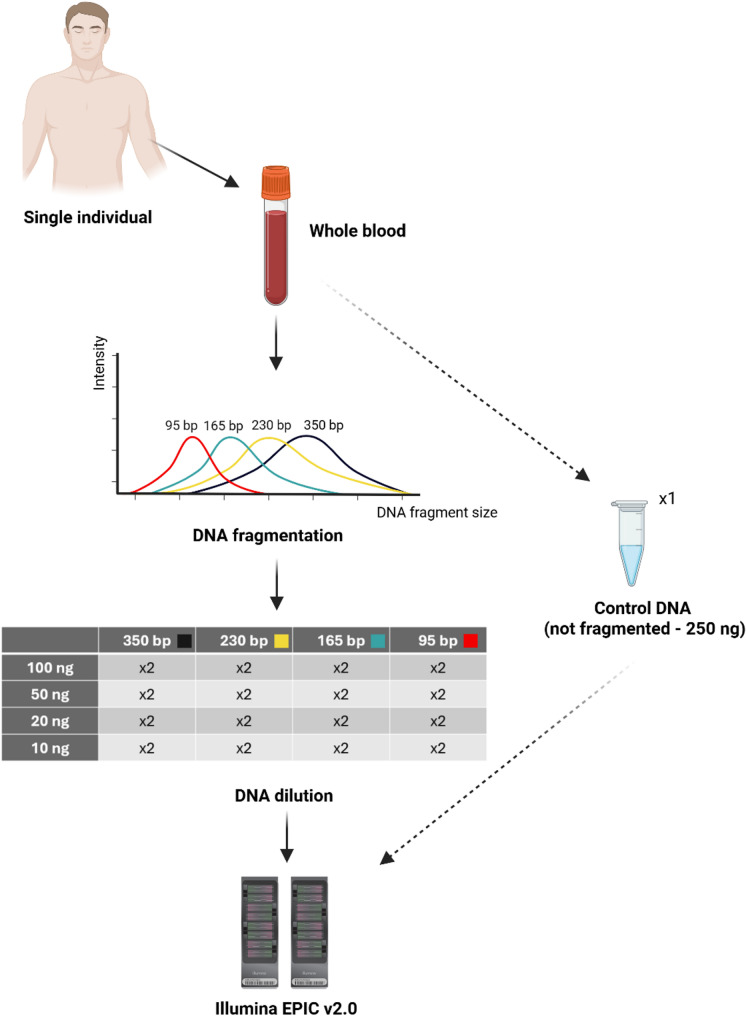
Background: The HumanMethylationEPIC v2.0 BeadChip (EPIC v2.0) microarray is a widely used tool for genome-wide DNA methylation (DNAm) analysis, designed for high-quality human DNA with a recommended input of 250 ng. However, in clinical and forensic settings, DNA samples may be of low quality and/or quantity (highly fragmented and/or available in low amounts). This study assessed the performance of the EPIC v2.0 on DNA samples with various combinations of average DNA fragment size (350, 230, 165, and 95 bp) and DNA input amount (100, 50, 20, and 10 ng), compared to a paired control sample analyzed under optimal conditions (high-quality DNA and 250 ng DNA input).
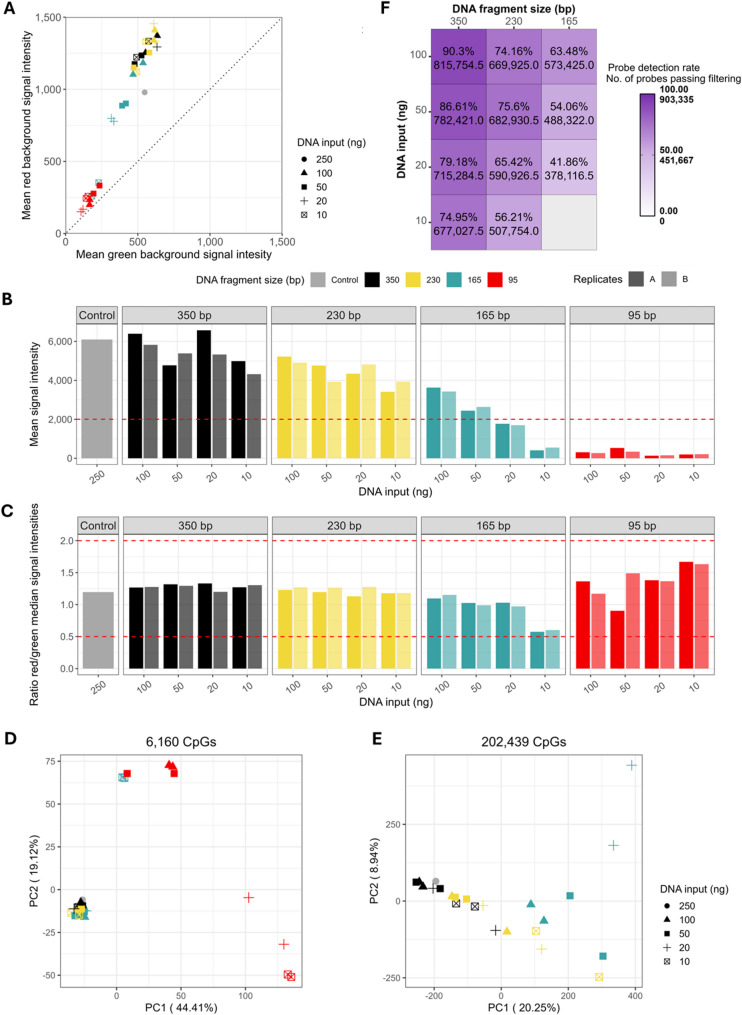
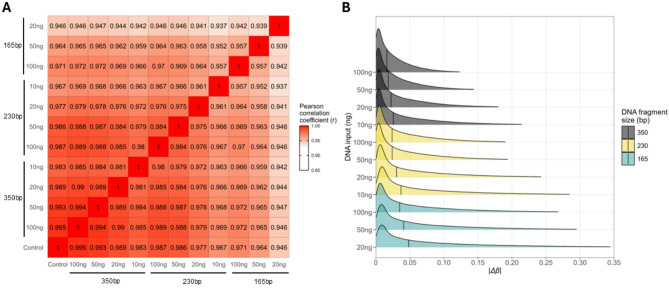
Results: The best performance was obtained for samples with average DNA fragment size of 350 bp and 100 ng DNA input (~ 90% probe detection rate, r = 0.995, and median absolute beta value differences|Δβ| = 0.012 when compared with the control sample). Samples with lower average DNA fragment sizes and DNA input amount performed worse, with the lowest probe detection rate (~ 43%), r = 0.946, and the highest|Δβ| (0.038). Samples with average DNA fragment sizes of 95 bp and those with 165 bp at 10 ng DNA input failed to pass sample quality control (QC). CpG sites with intermediate DNAm values (β = 0.1-0.9) showed higher|Δβ| than the extreme DNAm values (β = 0-0.1, and β = 0.9-1). Finally, we assessed an application of DNAm by performing epigenetic age analysis, and observed mean absolute errors (MAEs) below 10 years for 350 bp samples across four epigenetic clocks.
Conclusions: Both DNA fragment size and DNA input amounts affect DNAm analysis on the EPIC v2.0, with the investigated DNA fragment size having a greater impact than the investigated DNA input amount. DNAm measurements were achieved with the EPIC v2.0 microarray down to an average DNA fragment size of 165 bp and a 20 ng DNA input. Highly fragmented DNA (95 bp) did not result in usable DNAm analysis as all samples failed QC. Overall, our study demonstrates the potential and limitations of EPIC v2.0 microarray with low quality and quantity DNA samples.
期刊介绍:
iological Procedures Online publishes articles that improve access to techniques and methods in the medical and biological sciences.
We are also interested in short but important research discoveries, such as new animal disease models.
Topics of interest include, but are not limited to:
Reports of new research techniques and applications of existing techniques
Technical analyses of research techniques and published reports
Validity analyses of research methods and approaches to judging the validity of research reports
Application of common research methods
Reviews of existing techniques
Novel/important product information
Biological Procedures Online places emphasis on multidisciplinary approaches that integrate methodologies from medicine, biology, chemistry, imaging, engineering, bioinformatics, computer science, and systems analysis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


