First-principles computations on the structural, elastic, mechanical, electronic, optical, and thermal properties of doped halide perovskites CsSnCl3-xBrx (x = 0, 1, 2, 3) with cubic and tetragonal symmetries: A (DFT-GGA-mBJ)-based investigation
IF 2.4
3区 化学
Q4 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
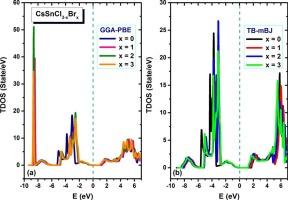
Doped halide perovskites CsSnCl3-xBrx (x = 0, 1, 2, 3) with semiconductor nature are promising inorganic materials for solar cells and photovoltaics and other optoelectronic devices, having high absorption and structural stability and less toxicity. Herein, we have investigated the structural, elastic, mechanical, thermal, optical, and electronic properties of CsSnCl3-xBrx using GGA-PBE and TB-mBJ approximations. The crystal structure of CsSnCl3-xBrx with cubic (Pm-3 m) and tetragonal (P4/mmm) symmetries depends on the x value, which influences their essential physical properties. The computed lattice constants are in good agreement with the previous reports. The results obtained in this study show that all these CsSnCl3-xBrx have semiconductor nature with a direct band gap that lies in the visible range ( = 0.895–1.749 eV). All compounds of CsSnCl3-xBrx are mechanically stable with inherent ductility and a Debye temperature of 97.4–171.1 K. Also, the optical computations reveal high absorption power in the visible-ultraviolet range.
掺杂卤化物钙钛矿CsSnCl3-xBrx (x = 0,1,2,3)的结构、弹性、力学、电子、光学和热性质的第一性原理计算:基于A (DFT-GGA-mBJ)的研究
掺杂卤化物钙钛矿CsSnCl3-xBrx (x = 0,1,2,3)具有半导体性质,是太阳能电池和光伏等光电器件中很有前途的无机材料,具有较高的吸收率和结构稳定性,毒性小。在此,我们使用GGA-PBE和TB-mBJ近似研究了CsSnCl3-xBrx的结构、弹性、力学、热、光学和电子性能。具有立方(pm - 3m)和四方(P4/mmm)对称性的CsSnCl3-xBrx晶体结构取决于x值,x值影响其基本物理性质。计算得到的晶格常数与前人的报道基本一致。本研究结果表明,这些CsSnCl3-xBrx均具有半导体性质,其直接带隙位于可见光范围内(Eg = 0.895-1.749 eV)。所有CsSnCl3-xBrx化合物均具有机械稳定性,具有固有的延展性,德拜温度θD为97.4-171.1 K。此外,光学计算表明在可见-紫外范围内具有较高的吸收能力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Chemical Physics
化学-物理:原子、分子和化学物理
CiteScore
4.60
自引率
4.30%
发文量
278
审稿时长
39 days
期刊介绍:
Chemical Physics publishes experimental and theoretical papers on all aspects of chemical physics. In this journal, experiments are related to theory, and in turn theoretical papers are related to present or future experiments. Subjects covered include: spectroscopy and molecular structure, interacting systems, relaxation phenomena, biological systems, materials, fundamental problems in molecular reactivity, molecular quantum theory and statistical mechanics. Computational chemistry studies of routine character are not appropriate for this journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


