{"title":"Statistical Approach to the Free-Energy Diagram of the Nitrogen Reduction Reaction on Mo2C MXene","authors":"Diwakar Singh, Ebrahim Tayyebi, Kai S. Exner","doi":"10.1002/celc.202500196","DOIUrl":null,"url":null,"abstract":"<p>Accurate free-energy landscapes are essential for understanding electrocatalytic processes, especially those involving proton–coupled electron transfer. While density functional theory (DFT) is widely used to model such reactions, it often introduces significant errors in the computed free energies of gas-phase reference molecules, leading to inconsistencies in the derivation of the free-energy changes of the elementary reaction steps. This study presents and compares different correction schemes to address gas-phase DFT errors. Unlike conventional methods that rely on bond–order–based adjustments, this approach reconstructs the formation free energy of target molecules as a linear combination of theoretically determined formation free energies of carefully selected reference molecules. This framework ensures consistency across the reaction network while avoiding dependence on the bond order. This methodology applies to the nitrogen reduction reaction on Mo<sub>2</sub>C(0001) MXene using dispersion–corrected DFT calculations. The incorporation of gas-phase corrections significantly reshapes the free-energy profile and alters catalytic activity descriptors, including the largest free-energy span of the <i>G</i><sub>max</sub>(<i>U</i>) descriptor. Findings highlight the importance of thermodynamic accuracy in computational electrocatalysis and provide a generalizable framework that improves the reliability of DFT-based predictions across a wide range of electrochemical systems for energy conversion and storage.</p>","PeriodicalId":142,"journal":{"name":"ChemElectroChem","volume":"12 17","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2025-08-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://chemistry-europe.onlinelibrary.wiley.com/doi/epdf/10.1002/celc.202500196","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ChemElectroChem","FirstCategoryId":"92","ListUrlMain":"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/celc.202500196","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ELECTROCHEMISTRY","Score":null,"Total":0}
引用次数: 0
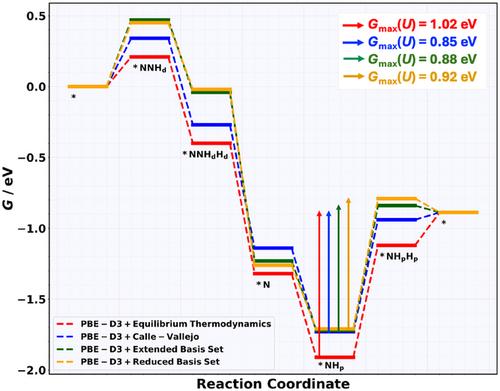
Abstract
Accurate free-energy landscapes are essential for understanding electrocatalytic processes, especially those involving proton–coupled electron transfer. While density functional theory (DFT) is widely used to model such reactions, it often introduces significant errors in the computed free energies of gas-phase reference molecules, leading to inconsistencies in the derivation of the free-energy changes of the elementary reaction steps. This study presents and compares different correction schemes to address gas-phase DFT errors. Unlike conventional methods that rely on bond–order–based adjustments, this approach reconstructs the formation free energy of target molecules as a linear combination of theoretically determined formation free energies of carefully selected reference molecules. This framework ensures consistency across the reaction network while avoiding dependence on the bond order. This methodology applies to the nitrogen reduction reaction on Mo2C(0001) MXene using dispersion–corrected DFT calculations. The incorporation of gas-phase corrections significantly reshapes the free-energy profile and alters catalytic activity descriptors, including the largest free-energy span of the Gmax(U) descriptor. Findings highlight the importance of thermodynamic accuracy in computational electrocatalysis and provide a generalizable framework that improves the reliability of DFT-based predictions across a wide range of electrochemical systems for energy conversion and storage.
期刊介绍:
ChemElectroChem is aimed to become a top-ranking electrochemistry journal for primary research papers and critical secondary information from authors across the world. The journal covers the entire scope of pure and applied electrochemistry, the latter encompassing (among others) energy applications, electrochemistry at interfaces (including surfaces), photoelectrochemistry and bioelectrochemistry.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


