Ruminococcus gnavus and Biofilm Markers in Feces From Primary Bile Acid Diarrhea Patients Indicate New Disease Mechanisms and Potential for Diagnostic Testing
Evette B.M. Hillman , Danielle Carson , Julian R.F. Walters , Martin Fritzsche , Ryan Mate , Katie E. Chappell , Elena Chekmeneva , Maria Gomez Romero , Stephen J. Lewis , Sjoerd Rijpkema , Elizabeth M.H. Wellington , Ramesh Arasaradnam , Gregory C.A. Amos
{"title":"Ruminococcus gnavus and Biofilm Markers in Feces From Primary Bile Acid Diarrhea Patients Indicate New Disease Mechanisms and Potential for Diagnostic Testing","authors":"Evette B.M. Hillman , Danielle Carson , Julian R.F. Walters , Martin Fritzsche , Ryan Mate , Katie E. Chappell , Elena Chekmeneva , Maria Gomez Romero , Stephen J. Lewis , Sjoerd Rijpkema , Elizabeth M.H. Wellington , Ramesh Arasaradnam , Gregory C.A. Amos","doi":"10.1016/j.gastha.2025.100712","DOIUrl":null,"url":null,"abstract":"<div><h3>Background and Aims</h3><div>Bile acid diarrhea (BAD) is a common cause of frequent loose stools, urgency, and incontinence, which is under-recognized due to limited diagnostic test availability and unclear pathogenesis. This study aimed to investigate fecal changes in well-defined subjects.</div></div><div><h3>Methods</h3><div>Fecal samples were compared in BAD patients (n = 26), diagnosed by SeHCAT testing, and healthy controls (n = 21). Shotgun metagenomic sequencing was used to identify microbiome species and functional genes. An extended set of 38 bile acids was quantified by liquid chromatography mass spectrometry, including various epimers and intermediates, such as iso- (3-beta-OH), oxo (keto), allo (5-alpha), and 3-sulfated forms.</div></div><div><h3>Results</h3><div>Alpha diversity, reflecting microbial richness, was reduced in BAD patients with severe forms of the disease, while beta diversity demonstrated distinct microbial profiles between groups. <em>Ruminococcus gnavus</em> (<em>R. gnavus</em>) was prevalent in BAD patients but rare in controls (odds ratio = 73), while <em>Firmicutes bacterium</em> CAG110, <em>Eubacterium siraeum</em> and 2 <em>Oscillibacter</em> species were less common in BAD (odds ratios = 25–30). Overall, 99 taxa differed significantly between groups. Bile acidtransforming genes (<em>baiA</em>, <em>baiB</em>, <em>hdhA</em>) were more abundant in BAD samples (<em>P</em> ≤ .0012). Most fecal bile acids, including iso-bile acids and intermediates, were higher in BAD. Elevated ursodeoxycholic acid-3-sulfate and relatively lower lithocholic acid and allo-bile acids, including isoallolithocholic acid, reflect changes in bacterial metabolism. Biofilm-associated genes (<em>bssS</em>, <em>pgaA</em>, <em>pgaB</em>) were markedly elevated in BAD patients (<em>P</em> ≤ .00008). SeHCAT values negatively correlated with <em>R. gnavus</em> (rho −0.53, <em>P</em> = .008) and positively with <em>E</em><em>ubacterium siraeum</em> (rho 0.41, <em>P</em> = .041).</div></div><div><h3>Conclusion</h3><div>BAD may result from an overgrowth of <em>R. gnavus</em>, associated with intestinal biofilms and an altered bile acid metabolism.</div></div>","PeriodicalId":73130,"journal":{"name":"Gastro hep advances","volume":"4 9","pages":"Article 100712"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Gastro hep advances","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2772572325000998","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background and Aims
Bile acid diarrhea (BAD) is a common cause of frequent loose stools, urgency, and incontinence, which is under-recognized due to limited diagnostic test availability and unclear pathogenesis. This study aimed to investigate fecal changes in well-defined subjects.
Methods
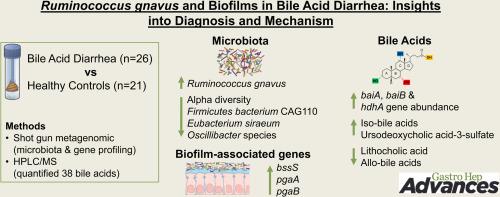
Fecal samples were compared in BAD patients (n = 26), diagnosed by SeHCAT testing, and healthy controls (n = 21). Shotgun metagenomic sequencing was used to identify microbiome species and functional genes. An extended set of 38 bile acids was quantified by liquid chromatography mass spectrometry, including various epimers and intermediates, such as iso- (3-beta-OH), oxo (keto), allo (5-alpha), and 3-sulfated forms.
Results
Alpha diversity, reflecting microbial richness, was reduced in BAD patients with severe forms of the disease, while beta diversity demonstrated distinct microbial profiles between groups. Ruminococcus gnavus (R. gnavus) was prevalent in BAD patients but rare in controls (odds ratio = 73), while Firmicutes bacterium CAG110, Eubacterium siraeum and 2 Oscillibacter species were less common in BAD (odds ratios = 25–30). Overall, 99 taxa differed significantly between groups. Bile acidtransforming genes (baiA, baiB, hdhA) were more abundant in BAD samples (P ≤ .0012). Most fecal bile acids, including iso-bile acids and intermediates, were higher in BAD. Elevated ursodeoxycholic acid-3-sulfate and relatively lower lithocholic acid and allo-bile acids, including isoallolithocholic acid, reflect changes in bacterial metabolism. Biofilm-associated genes (bssS, pgaA, pgaB) were markedly elevated in BAD patients (P ≤ .00008). SeHCAT values negatively correlated with R. gnavus (rho −0.53, P = .008) and positively with Eubacterium siraeum (rho 0.41, P = .041).
Conclusion
BAD may result from an overgrowth of R. gnavus, associated with intestinal biofilms and an altered bile acid metabolism.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


