Jason Sardell, Sayoni Das, Gert Lykke Moeller, Marianna Sanna, Karolina Chocian, Krystyna Taylor, Andy Malinowski, Colin Stubberfield, Amy Rochlin, Steve Gardner
{"title":"Identification and Validation of Novel Combinatorial Genetic Risk Factors for Endometriosis across Multiple UK and US Patient Cohorts.","authors":"Jason Sardell, Sayoni Das, Gert Lykke Moeller, Marianna Sanna, Karolina Chocian, Krystyna Taylor, Andy Malinowski, Colin Stubberfield, Amy Rochlin, Steve Gardner","doi":"10.1101/2025.08.13.25333595","DOIUrl":null,"url":null,"abstract":"<p><p>Background Endometriosis affects about 10% of women usually of reproductive age. It often has severe negative impacts on patients' quality of life, but the average time to a definitive diagnosis remains 7-9 years, and there are few effective therapeutic options. Relatively little is known about the genetic drivers of the disease even though its heritability is fairly high. A recent large genome wide association study (GWAS) meta-analysis identified 42 genomic loci associated with risk of endometriosis, but together these explain only 5% of disease variance. Methods We used the PrecisionLife combinatorial analytics platform to identify multi-SNP disease signatures significantly associated with endometriosis in a white European UK Biobank (UKB) cohort. We assessed the reproducibility of these multi-SNP disease signatures as well as 35 of the 42 meta-GWAS SNPs in a multi-ancestry American endometriosis cohort from All of Us (AoU) after controlling for population structure. Results We identified 1,709 disease signatures, comprising 2,957 unique SNPs in combinations of 2-5 SNPs, that were associated with increased prevalence of endometriosis in UKB. Pathways enriched in the disease signatures included cell adhesion, proliferation and migration, cytoskeleton remodeling, angiogenesis as well as biological processes involved in fibrosis and neuropathic pain. We observed a significant enrichment of these signatures (58-88%, p<0.04) that are also positively associated with endometriosis in the AoU cohort, including one 2-SNP signature that is individually significant. Reproducibility rates were greatest for higher frequency signatures, ranging from 80-88% for signatures with greater than 9% frequency (p<0.01) in AoU. Encouragingly, the disease signatures also show high reproducibility rates in non-white European AoU sub-cohorts (66-76%, p<0.04 for signatures with greater than 4% frequency). A total of 195 unique SNPs mapping to 98 genes were identified in the high frequency reproducing signatures (>9%). Of these, 7 genes were previously identified in the endometriosis meta-GWAS study and 16 genes have a previous association with endometriosis. 75 novel genes were identified in this study. We characterized 9 novel genes that occur at the highest frequency in reproducing signatures and that do not contain any SNPs linked to known GWAS genes, providing new evidence for links between endometriosis and autophagy and macrophage biology. Reproducibility rates, ranging between 73% to 85%. are especially strong for the signatures that contain these 9 genes independently of any SNPs mapping to the meta-GWAS genes. Conclusion Although using much smaller, less well-characterized datasets than the previous whole genome meta-GWAS study, combinatorial analysis has provided important new insights into the genetics and biology of endometriosis including reproducible biologically relevant genes that are overlooked by GWAS approaches. The 75 novel gene associations provide new insights and routes for study of the disease and potential new therapies. Several of the novel genes identified are credible targets for drug discovery, repurposing and/or repositioning. Using the disease signatures identified as genetic biomarkers in trials of candidates drugs targeting specific mechanisms will enable precision medicine-based approaches. We hope this will encourage new targeted therapy discovery efforts.</p>","PeriodicalId":94281,"journal":{"name":"medRxiv : the preprint server for health sciences","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12363715/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"medRxiv : the preprint server for health sciences","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2025.08.13.25333595","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background Endometriosis affects about 10% of women usually of reproductive age. It often has severe negative impacts on patients' quality of life, but the average time to a definitive diagnosis remains 7-9 years, and there are few effective therapeutic options. Relatively little is known about the genetic drivers of the disease even though its heritability is fairly high. A recent large genome wide association study (GWAS) meta-analysis identified 42 genomic loci associated with risk of endometriosis, but together these explain only 5% of disease variance. Methods We used the PrecisionLife combinatorial analytics platform to identify multi-SNP disease signatures significantly associated with endometriosis in a white European UK Biobank (UKB) cohort. We assessed the reproducibility of these multi-SNP disease signatures as well as 35 of the 42 meta-GWAS SNPs in a multi-ancestry American endometriosis cohort from All of Us (AoU) after controlling for population structure. Results We identified 1,709 disease signatures, comprising 2,957 unique SNPs in combinations of 2-5 SNPs, that were associated with increased prevalence of endometriosis in UKB. Pathways enriched in the disease signatures included cell adhesion, proliferation and migration, cytoskeleton remodeling, angiogenesis as well as biological processes involved in fibrosis and neuropathic pain. We observed a significant enrichment of these signatures (58-88%, p<0.04) that are also positively associated with endometriosis in the AoU cohort, including one 2-SNP signature that is individually significant. Reproducibility rates were greatest for higher frequency signatures, ranging from 80-88% for signatures with greater than 9% frequency (p<0.01) in AoU. Encouragingly, the disease signatures also show high reproducibility rates in non-white European AoU sub-cohorts (66-76%, p<0.04 for signatures with greater than 4% frequency). A total of 195 unique SNPs mapping to 98 genes were identified in the high frequency reproducing signatures (>9%). Of these, 7 genes were previously identified in the endometriosis meta-GWAS study and 16 genes have a previous association with endometriosis. 75 novel genes were identified in this study. We characterized 9 novel genes that occur at the highest frequency in reproducing signatures and that do not contain any SNPs linked to known GWAS genes, providing new evidence for links between endometriosis and autophagy and macrophage biology. Reproducibility rates, ranging between 73% to 85%. are especially strong for the signatures that contain these 9 genes independently of any SNPs mapping to the meta-GWAS genes. Conclusion Although using much smaller, less well-characterized datasets than the previous whole genome meta-GWAS study, combinatorial analysis has provided important new insights into the genetics and biology of endometriosis including reproducible biologically relevant genes that are overlooked by GWAS approaches. The 75 novel gene associations provide new insights and routes for study of the disease and potential new therapies. Several of the novel genes identified are credible targets for drug discovery, repurposing and/or repositioning. Using the disease signatures identified as genetic biomarkers in trials of candidates drugs targeting specific mechanisms will enable precision medicine-based approaches. We hope this will encourage new targeted therapy discovery efforts.
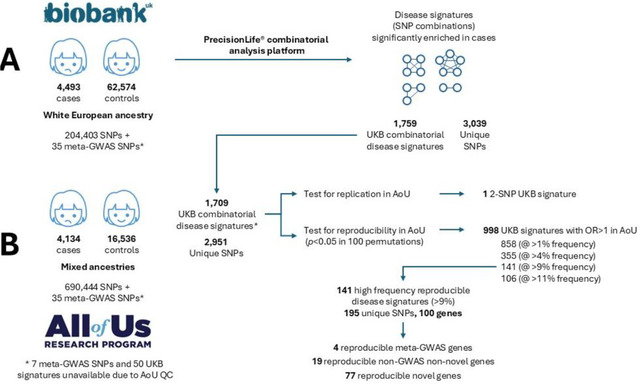
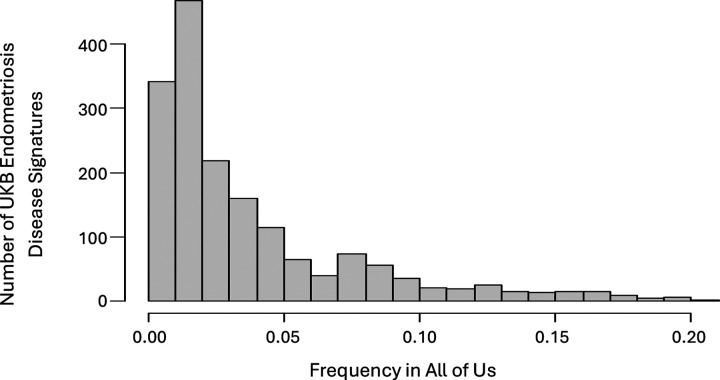
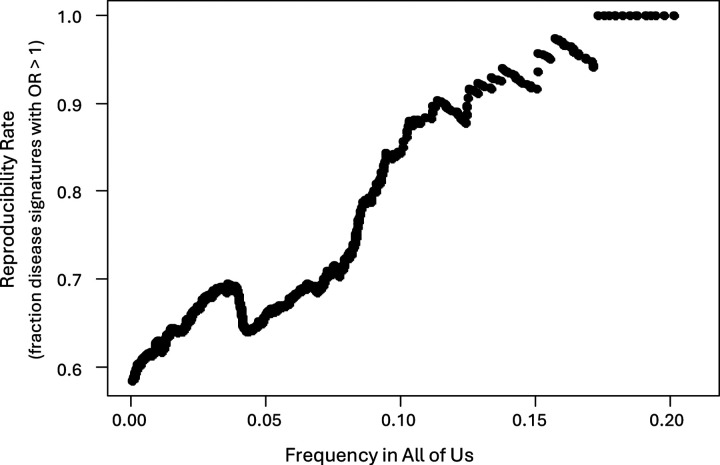
背景:子宫内膜异位症影响约10%的育龄妇女。它通常会对患者的生活质量产生严重的负面影响,但确诊的平均时间仍为7-9年,而且几乎没有有效的治疗选择。尽管这种疾病的遗传率相当高,但人们对这种疾病的遗传驱动因素所知相对较少。最近的一项大型基因组全关联研究(GWAS)荟萃分析确定了42个与子宫内膜异位症风险相关的基因组位点,但这些位点加起来只能解释5%的疾病变异。方法:我们使用PrecisionLife®组合分析平台在欧洲英国生物银行(UKB)白人队列中识别与子宫内膜异位症显著相关的多snp疾病特征。在控制人群结构后,我们评估了这些多snp疾病特征的可重复性,以及最近一项meta-GWAS研究在来自All of Us (AoU)的多祖先美国子宫内膜异位症队列中发现的42个snp中的35个。结果:我们确定了1,709种疾病特征,包括2,957个独特的snp(2-5个snp的组合),这些特征与UKB子宫内膜异位症的患病率增加有关。我们观察到这些特征显著富集(58-88%,p - p - 9%)。其中,4个基因先前在子宫内膜异位症meta-GWAS研究中被确定,19个基因先前在OpenTargets 1中与子宫内膜异位症相关。本研究共鉴定出77个新基因。我们鉴定了9个在复制特征中出现频率最高的新基因,这些基因不包含任何与已知GWAS基因相关的snp,为子宫内膜异位症与自噬和巨噬细胞生物学之间的联系提供了新的证据。重现率在73% ~ 85%之间。对于包含这9个基因的独立于任何指向meta-GWAS基因的snp的特征尤其强烈。这些基因还包括一些对子宫内膜异位症具有可靠的治疗发现,重新利用和/或重新定位潜力的新靶点。结论:虽然使用的数据集比之前的全基因组meta-GWAS研究要小得多,特征也不完善,但组合分析为子宫内膜异位症的遗传学和生物学提供了重要的新见解。在一个独立的、祖先多样化的数据集中发现了77个新的高频基因关联,这表明组合分析可以识别被GWAS方法忽视的生物学相关基因。这些新基因中的一些将成为药物发现和重新利用的可靠靶点,如突出显示的例子所示。跨数据集和谱系结果的广泛可重复性表明,组合疾病特征可用于识别不同的机制病因,这些病因有可能为基于精确医学的方法提供信息,并为这种复杂疾病产生新的临床治疗方法。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


