A comparison of 27 Arabidopsis thaliana genomes and the path toward an unbiased characterization of genetic polymorphism
IF 29
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
Abstract
Making sense of whole-genome polymorphism data is challenging, but it is essential for overcoming the biases in SNP data. Here we analyze 27 genomes of Arabidopsis thaliana to illustrate these issues. Genome size variation is mostly due to tandem repeat regions that are difficult to assemble. However, while the rest of the genome varies little in length, it is full of structural variants, mostly due to transposon insertions. Because of this, the pangenome coordinate system grows rapidly with sample size and ultimately becomes 70% larger than the size of any single genome, even for n = 27. Finally, we show how short-read data are biased by read mapping. SNP calling is biased by the choice of reference genome, and both transcriptome and methylome profiling results are affected by mapping reads to a reference genome rather than to the genome of the assayed individual. New concepts for comparing the genomes of 27 naturally inbred Arabidopsis thaliana accessions provide essential insights into obtaining a less biased view of whole-genome polymorphism.
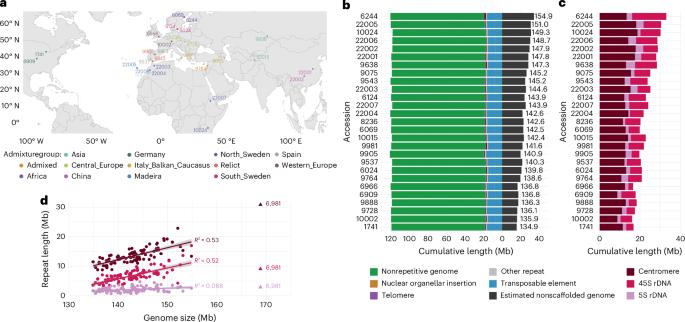
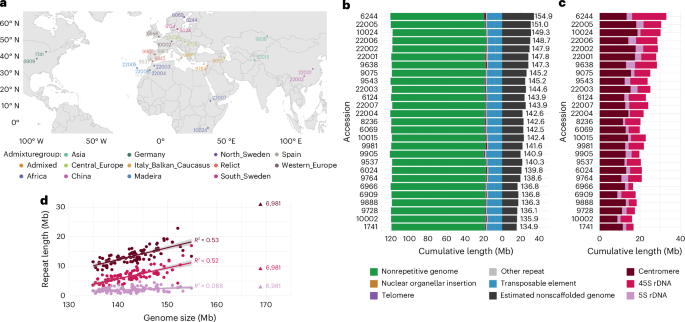
27个拟南芥基因组的比较及其对遗传多态性的无偏定性。
理解全基因组多态性数据具有挑战性,但对于克服SNP数据的偏差至关重要。在这里,我们分析27个拟南芥基因组来说明这些问题。基因组大小的变化主要是由于串联重复区域难以组装。然而,虽然基因组的其余部分长度变化不大,但它充满了结构变异,主要是由于转座子插入。正因为如此,泛基因组坐标系统随着样本量的增加而迅速增长,最终比任何单个基因组的大小都要大70%,即使n = 27也是如此。最后,我们展示了短读数据是如何通过读映射产生偏差的。SNP调用受参考基因组选择的影响,转录组和甲基组分析结果都受到参考基因组而不是被测个体基因组的定位读数的影响。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


