{"title":"Identification of three novel GNAO1 variants in a Chinese cohort with GNAO1 encephalopathy: expanding the clinical and genetic spectrum.","authors":"Daoqi Mei, Yu Gu, Bingbing Zhang, Shiyue Mei, Xiaona Wang, Yuanning Ma, Jie Deng, Jihong Tang","doi":"10.1186/s13023-025-03984-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>To summarize the clinical characteristics of a cohort of nine Chinese children with GNAO1 encephalopathy and analyze their genotypes.</p><p><strong>Methods: </strong>A retrospective study was conducted on nine children diagnosed with GNAO1 encephalopathy at the Neurology Department of two children's hospitals between January 2019 and December 2022. Their clinical manifestations, genetic test results, cranial imaging, electroencephalography and treatment were summarized. Their prognosis was followed up.</p><p><strong>Results: </strong>All nine patients presented with moderate-to-severe psychomotor developmental delay and dystonia. Six patients exhibited neonatal or infantile-onset epilepsy, manifesting as generalized tonic-clonic seizure, myoclonic seizure, epileptic spasms, and were diagnosed with developmental and epileptic encephalopathy 17 (DEE 17). Two patients presented with choreoathetosis in infancy without epileptic seizure and were diagnosed with the neurodevelopmental disorder with involuntary movements (NEDIM). One patient presented with choreoathetosis at two years of age and developed focal seizures at six years of age, representing an intermediate phenotype. During a follow-up period of 0.8-3.5 years, one child died due to infection. The remaining eight continued to exhibit psychomotor retardation. Pathogenic or likely pathogenic de novo heterozygous missense variants in GNAO1 were identified in all nine cases. Among these, the variants c.17G > T (p.Ser6Ile), c.119G > C (p.Gly40Ala), and c.748 C > T (p.Leu250Phe) are novel.</p><p><strong>Conclusion: </strong>In conclusion, we analyzed the clinical characteristics and genetic variants of a cohort of nine Chinese children with GNAO1 variants and identified three novel GNAO1 variants. Our study expanded the spectrum of genotypes and phenotypes in GNOA1-associated encephalopathy.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"440"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12363038/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03984-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Objective: To summarize the clinical characteristics of a cohort of nine Chinese children with GNAO1 encephalopathy and analyze their genotypes.
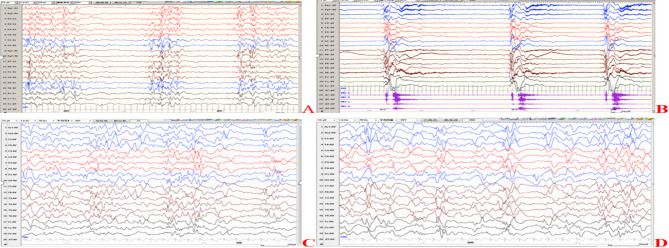
Methods: A retrospective study was conducted on nine children diagnosed with GNAO1 encephalopathy at the Neurology Department of two children's hospitals between January 2019 and December 2022. Their clinical manifestations, genetic test results, cranial imaging, electroencephalography and treatment were summarized. Their prognosis was followed up.
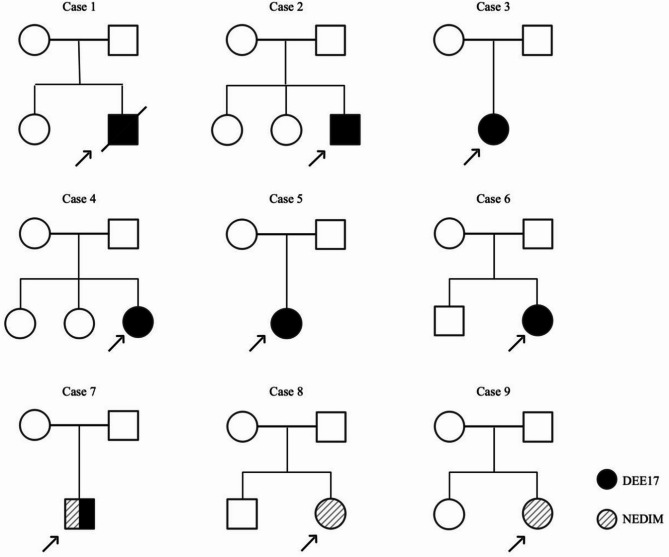
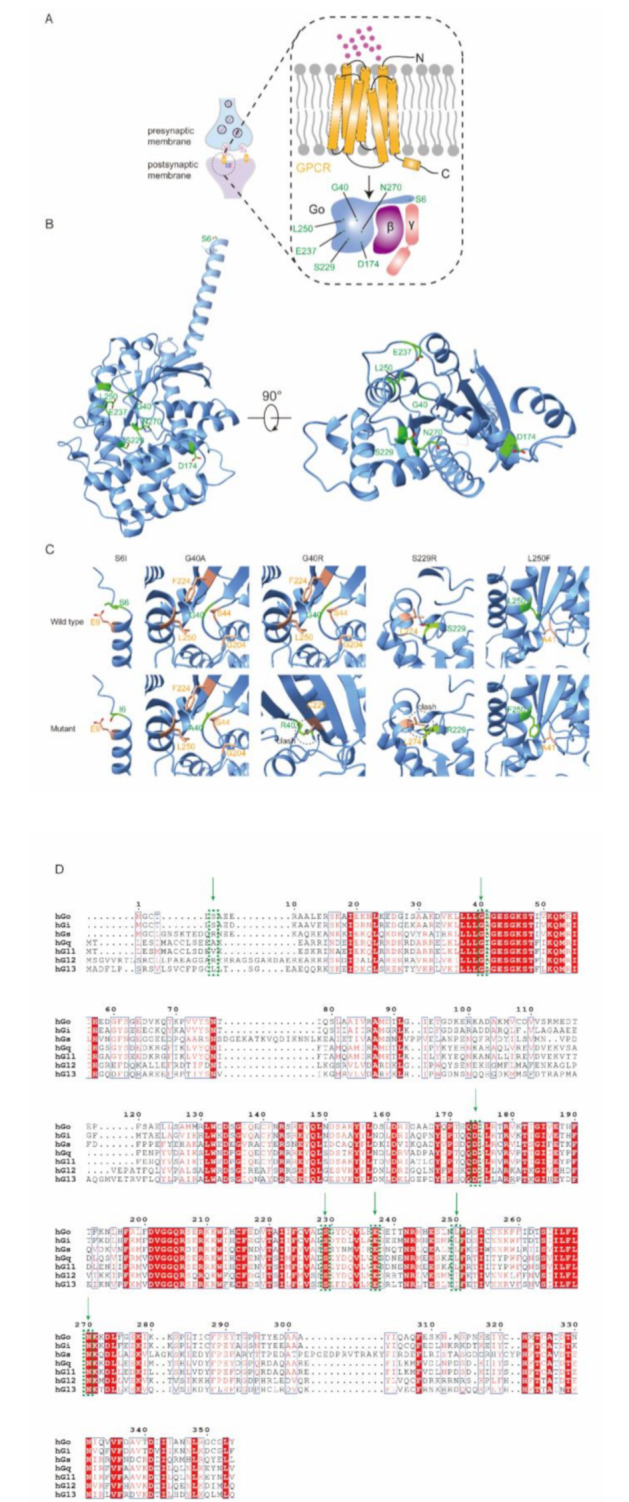
Results: All nine patients presented with moderate-to-severe psychomotor developmental delay and dystonia. Six patients exhibited neonatal or infantile-onset epilepsy, manifesting as generalized tonic-clonic seizure, myoclonic seizure, epileptic spasms, and were diagnosed with developmental and epileptic encephalopathy 17 (DEE 17). Two patients presented with choreoathetosis in infancy without epileptic seizure and were diagnosed with the neurodevelopmental disorder with involuntary movements (NEDIM). One patient presented with choreoathetosis at two years of age and developed focal seizures at six years of age, representing an intermediate phenotype. During a follow-up period of 0.8-3.5 years, one child died due to infection. The remaining eight continued to exhibit psychomotor retardation. Pathogenic or likely pathogenic de novo heterozygous missense variants in GNAO1 were identified in all nine cases. Among these, the variants c.17G > T (p.Ser6Ile), c.119G > C (p.Gly40Ala), and c.748 C > T (p.Leu250Phe) are novel.
Conclusion: In conclusion, we analyzed the clinical characteristics and genetic variants of a cohort of nine Chinese children with GNAO1 variants and identified three novel GNAO1 variants. Our study expanded the spectrum of genotypes and phenotypes in GNOA1-associated encephalopathy.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


