Chaoqun Zhu, Meimi Zhao, Luqi Zhao, Mingfu Wu, Yang K Xiang
{"title":"Cardiac β2 adrenergic receptor deletion drives calmodulin kinase II upregulation to induce connective tissue growth factor in cardiac fibrosis and diastolic dysfunction.","authors":"Chaoqun Zhu, Meimi Zhao, Luqi Zhao, Mingfu Wu, Yang K Xiang","doi":"10.1093/function/zqaf036","DOIUrl":null,"url":null,"abstract":"<p><p>Abnormalities of Ca2+ signaling in the heart lead to common cardiac remodeling in the pathogenesis of cardiovascular disorders. The activation of calmodulin-dependent protein kinase II (CaMKII) is regulated by elevated intracellular Ca2+ level in cardiomyocytes, driving the progression of myocardial dysfunction. In this study, using models of β2 adrenergic receptor (β2AR) deficiency in cardiomyocytes (β2AR-CKO), we observed an increased phosphorylation of CaMKII and upregulation of gene expression and protein level of the fibrotic marker connective tissue growth factor (CTGF) in the myocytes. In vivo treatment with the CaMKII inhibitor KN93 attenuated the upregulation of CTGF protein expression in β2AR-CKO hearts. Enhanced L-type calcium channel (LTCC) current was observed in β2AR-CKO cardiomyocytes following adrenergic stimulation, indicating a disruption of Ca2+ signaling. Treatment with the LTCC blocker nifedipine attenuated CaMKII activity and the expression of CTGF in β2AR-CKO hearts, confirming the upstream role of abnormal LTCC-Ca2+ signaling. Additionally, 8-month-old β2AR-CKO mice exhibited cardiac fibrosis and diastolic dysfunction. One month of in vivo nifedipine treatment improved both cardiac dysfunction and fibrosis in β2AR-CKO mice. These findings highlight the critical role of cardiomyocyte β2AR in maintaining LTCC-Ca2+ homeostasis. Loss of β2AR amplifies the Ca2+-CaMKII axis, promoting fibrosis and cardiomyopathy in aging hearts.</p>","PeriodicalId":73119,"journal":{"name":"Function (Oxford, England)","volume":" ","pages":""},"PeriodicalIF":3.8000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12448417/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Function (Oxford, England)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/function/zqaf036","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
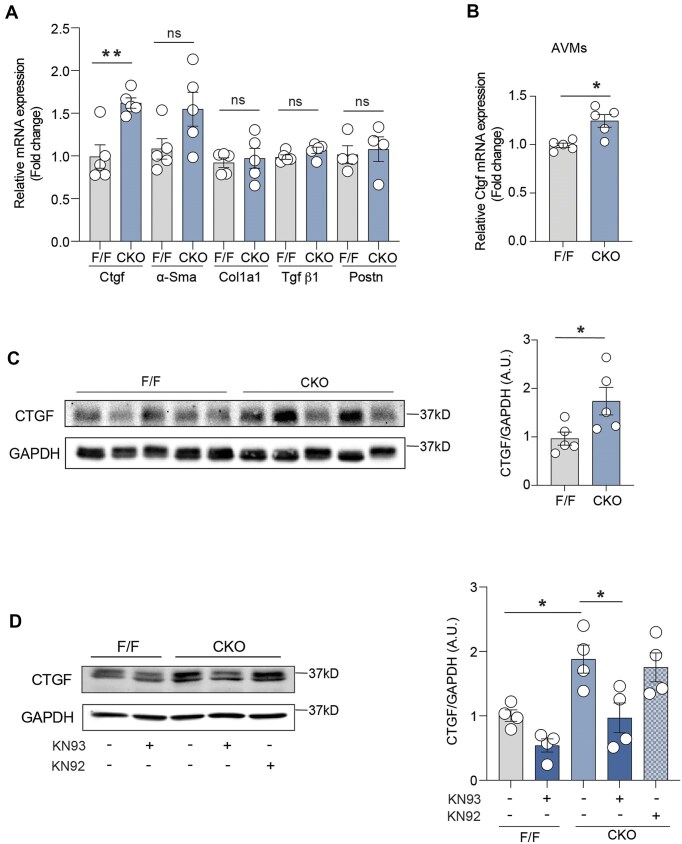
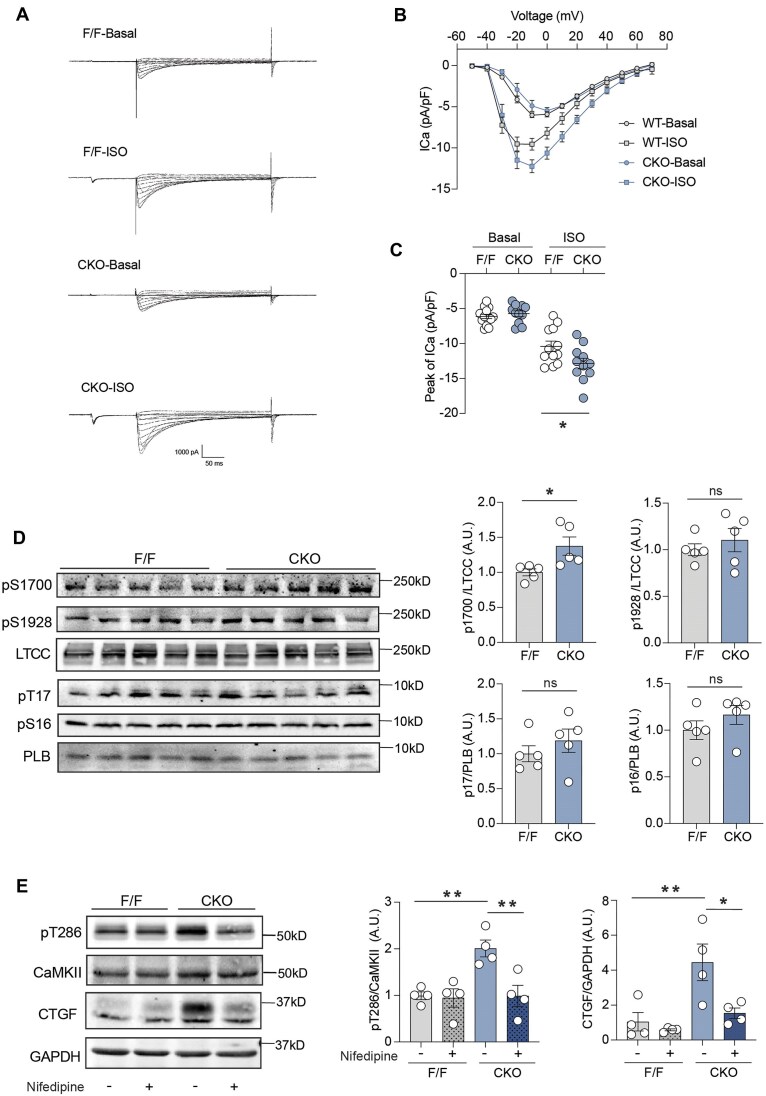
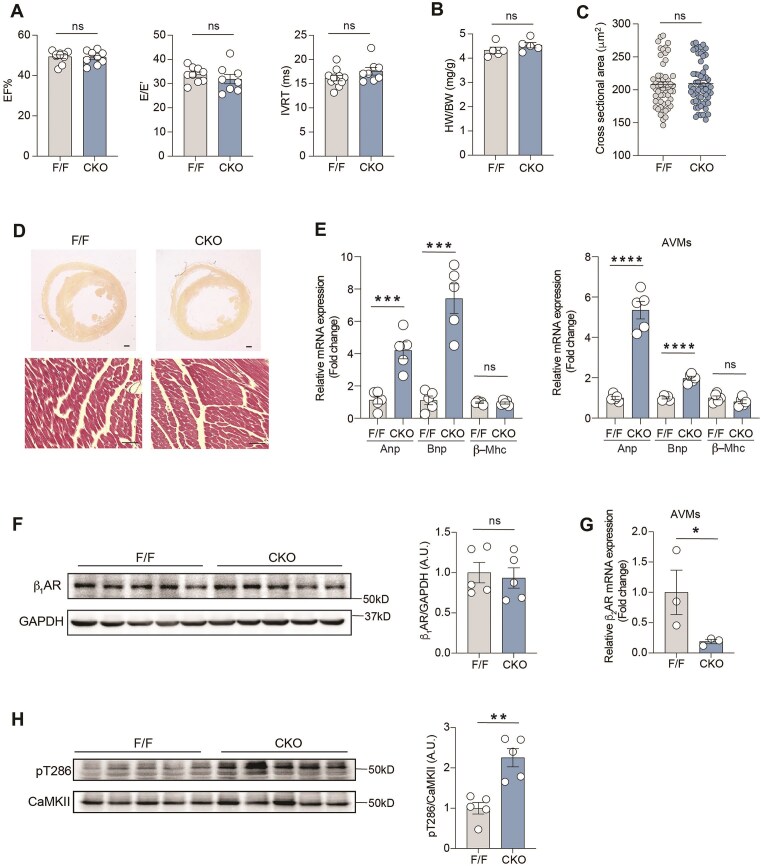
Abnormalities of Ca2+ signaling in the heart lead to common cardiac remodeling in the pathogenesis of cardiovascular disorders. The activation of calmodulin-dependent protein kinase II (CaMKII) is regulated by elevated intracellular Ca2+ level in cardiomyocytes, driving the progression of myocardial dysfunction. In this study, using models of β2 adrenergic receptor (β2AR) deficiency in cardiomyocytes (β2AR-CKO), we observed an increased phosphorylation of CaMKII and upregulation of gene expression and protein level of the fibrotic marker connective tissue growth factor (CTGF) in the myocytes. In vivo treatment with the CaMKII inhibitor KN93 attenuated the upregulation of CTGF protein expression in β2AR-CKO hearts. Enhanced L-type calcium channel (LTCC) current was observed in β2AR-CKO cardiomyocytes following adrenergic stimulation, indicating a disruption of Ca2+ signaling. Treatment with the LTCC blocker nifedipine attenuated CaMKII activity and the expression of CTGF in β2AR-CKO hearts, confirming the upstream role of abnormal LTCC-Ca2+ signaling. Additionally, 8-month-old β2AR-CKO mice exhibited cardiac fibrosis and diastolic dysfunction. One month of in vivo nifedipine treatment improved both cardiac dysfunction and fibrosis in β2AR-CKO mice. These findings highlight the critical role of cardiomyocyte β2AR in maintaining LTCC-Ca2+ homeostasis. Loss of β2AR amplifies the Ca2+-CaMKII axis, promoting fibrosis and cardiomyopathy in aging hearts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


