Benjamin X. Shi, Andrew S. Rosen, Tobias Schäfer, Andreas Grüneis, Venkat Kapil, Andrea Zen, Angelos Michaelides
{"title":"An accurate and efficient framework for modelling the surface chemistry of ionic materials","authors":"Benjamin X. Shi, Andrew S. Rosen, Tobias Schäfer, Andreas Grüneis, Venkat Kapil, Andrea Zen, Angelos Michaelides","doi":"10.1038/s41557-025-01884-y","DOIUrl":null,"url":null,"abstract":"<p>Quantum-mechanical simulations can offer atomic-level insights into chemical processes on surfaces that are crucial to advancing applications in heterogeneous catalysis, energy storage and greenhouse gas sequestration. Unfortunately, achieving the accuracy needed for reliable predictions has proven challenging. Density functional theory, widely used for its efficiency, can be inconsistent, necessitating accurate methods from correlated wavefunction theory. But high computational demands and substantial user intervention have traditionally made correlated wavefunction theory impractical to carry out for surfaces. Here we present an automated framework that leverages multilevel embedding approaches to apply correlated wavefunction theory to the surfaces of ionic materials with computational costs approaching those of density functional theory. With this framework, we reproduce experimental adsorption enthalpies for a diverse set of 19 adsorbate–surface systems. We further resolve debates on the adsorption configuration of several systems, while offering benchmarks to assess density functional theory. This framework is open source, facilitating the routine application of correlated wavefunction theory to complex problems involving the surfaces of ionic materials.</p><figure></figure>","PeriodicalId":18909,"journal":{"name":"Nature chemistry","volume":"18 1","pages":""},"PeriodicalIF":20.2000,"publicationDate":"2025-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1038/s41557-025-01884-y","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
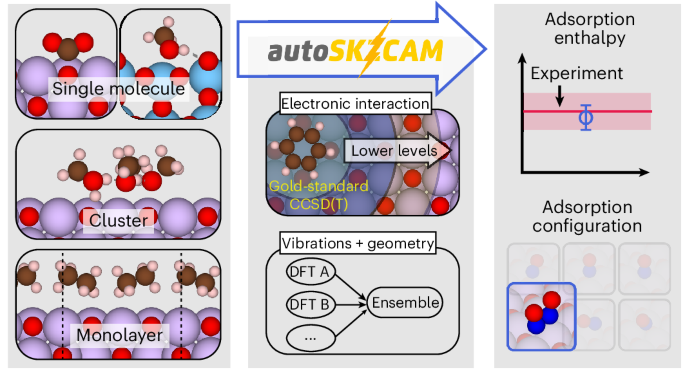
Quantum-mechanical simulations can offer atomic-level insights into chemical processes on surfaces that are crucial to advancing applications in heterogeneous catalysis, energy storage and greenhouse gas sequestration. Unfortunately, achieving the accuracy needed for reliable predictions has proven challenging. Density functional theory, widely used for its efficiency, can be inconsistent, necessitating accurate methods from correlated wavefunction theory. But high computational demands and substantial user intervention have traditionally made correlated wavefunction theory impractical to carry out for surfaces. Here we present an automated framework that leverages multilevel embedding approaches to apply correlated wavefunction theory to the surfaces of ionic materials with computational costs approaching those of density functional theory. With this framework, we reproduce experimental adsorption enthalpies for a diverse set of 19 adsorbate–surface systems. We further resolve debates on the adsorption configuration of several systems, while offering benchmarks to assess density functional theory. This framework is open source, facilitating the routine application of correlated wavefunction theory to complex problems involving the surfaces of ionic materials.
期刊介绍:
Nature Chemistry is a monthly journal that publishes groundbreaking and significant research in all areas of chemistry. It covers traditional subjects such as analytical, inorganic, organic, and physical chemistry, as well as a wide range of other topics including catalysis, computational and theoretical chemistry, and environmental chemistry.
The journal also features interdisciplinary research at the interface of chemistry with biology, materials science, nanotechnology, and physics. Manuscripts detailing such multidisciplinary work are encouraged, as long as the central theme pertains to chemistry.
Aside from primary research, Nature Chemistry publishes review articles, news and views, research highlights from other journals, commentaries, book reviews, correspondence, and analysis of the broader chemical landscape. It also addresses crucial issues related to education, funding, policy, intellectual property, and the societal impact of chemistry.
Nature Chemistry is dedicated to ensuring the highest standards of original research through a fair and rigorous review process. It offers authors maximum visibility for their papers, access to a broad readership, exceptional copy editing and production standards, rapid publication, and independence from academic societies and other vested interests.
Overall, Nature Chemistry aims to be the authoritative voice of the global chemical community.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


