{"title":"Bilateral Streak Ovaries in a Patient with Isochromosome Xq: A Case Report of a Turner Syndrome Variant.","authors":"Amadea Ivana Hartanto, Ruswana Anwar, Mirza Ismail, Erick Caesarrani Asmara, Putri Nadhira Adinda Adriansyah","doi":"10.2147/IMCRJ.S529460","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Turner syndrome (TS) is a chromosomal disorder characterized by complete or partial loss of one X chromosome. One structural variant, isochromosome Xq [46,X,i(Xq)], results in duplication of the long arm and loss of the short arm of the X chromosome (Xp), which contains genes essential for normal ovarian development and function. This chromosomal imbalance leads to accelerated germ cell apoptosis and subsequent ovarian dysfunction. In this case, a 33-year-old woman with chronic anovulation and uterine hypoplasia was diagnosed with 46,X,i(Xq) karyotype without evidence of mosaicism.</p><p><strong>Case illustration: </strong>A 33-year-old woman with a history of irregular menstruation since adolescence was referred for evaluation of uterine hypoplasia and chronic anovulation. Clinical findings included short stature, but no webbed neck or congenital heart defects, making this an atypical presentation of Turner syndrome. Transvaginal ultrasound revealed a small uterus and bilateral streak ovaries. Hormonal evaluation showed elevated FSH levels consistent with hypergonadotropic hypogonadism. Diagnostic laparoscopy confirmed bilateral streak ovaries with normal appearing uterus and fallopian tubes. Chromosomal analysis using G-banding revealed a 46,X,i(Xq) karyotype, indicating an isochromosome Xq abnormality, a recognized variant of Turner syndrome. This genetic alteration explains her ovarian dysfunction and infertility, highlighting the importance of chromosomal evaluation in cases of primary ovarian insufficiency.</p><p><strong>Conclusion: </strong>This case highlights a structurally abnormal but nonmosaic 46,X,i(Xq) karyotype variant of Turner syndrome presenting primarily with ovarian insufficiency. Despite the absence of classic phenotypic features such as webbed neck or congenital heart defects, a high index of suspicion led to the correct diagnosis. This case underscores the need to consider Turner syndrome variants in women with unexplained ovarian insufficiency, even in the absence of overt clinical stigmata, to guide appropriate genetic counseling and fertility planning.</p>","PeriodicalId":14337,"journal":{"name":"International Medical Case Reports Journal","volume":"18 ","pages":"953-961"},"PeriodicalIF":0.7000,"publicationDate":"2025-08-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12333654/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Medical Case Reports Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/IMCRJ.S529460","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
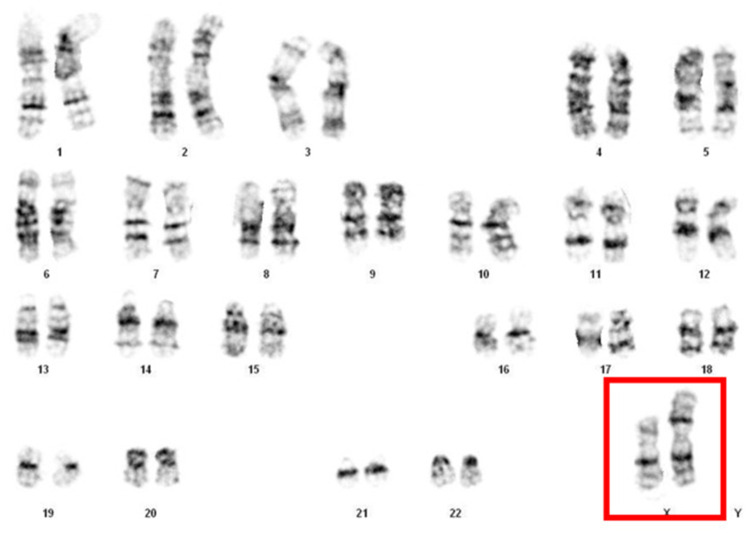
Introduction: Turner syndrome (TS) is a chromosomal disorder characterized by complete or partial loss of one X chromosome. One structural variant, isochromosome Xq [46,X,i(Xq)], results in duplication of the long arm and loss of the short arm of the X chromosome (Xp), which contains genes essential for normal ovarian development and function. This chromosomal imbalance leads to accelerated germ cell apoptosis and subsequent ovarian dysfunction. In this case, a 33-year-old woman with chronic anovulation and uterine hypoplasia was diagnosed with 46,X,i(Xq) karyotype without evidence of mosaicism.
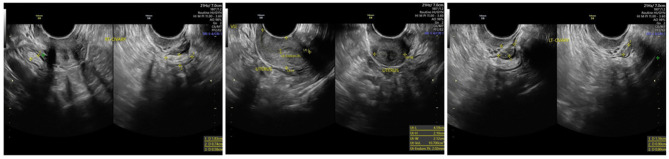
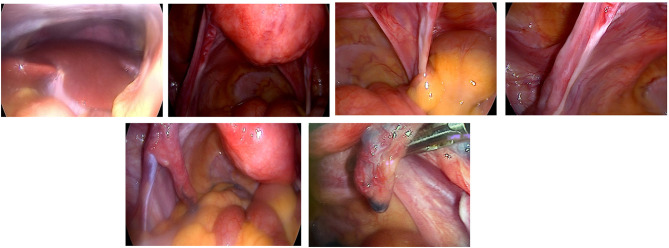
Case illustration: A 33-year-old woman with a history of irregular menstruation since adolescence was referred for evaluation of uterine hypoplasia and chronic anovulation. Clinical findings included short stature, but no webbed neck or congenital heart defects, making this an atypical presentation of Turner syndrome. Transvaginal ultrasound revealed a small uterus and bilateral streak ovaries. Hormonal evaluation showed elevated FSH levels consistent with hypergonadotropic hypogonadism. Diagnostic laparoscopy confirmed bilateral streak ovaries with normal appearing uterus and fallopian tubes. Chromosomal analysis using G-banding revealed a 46,X,i(Xq) karyotype, indicating an isochromosome Xq abnormality, a recognized variant of Turner syndrome. This genetic alteration explains her ovarian dysfunction and infertility, highlighting the importance of chromosomal evaluation in cases of primary ovarian insufficiency.
Conclusion: This case highlights a structurally abnormal but nonmosaic 46,X,i(Xq) karyotype variant of Turner syndrome presenting primarily with ovarian insufficiency. Despite the absence of classic phenotypic features such as webbed neck or congenital heart defects, a high index of suspicion led to the correct diagnosis. This case underscores the need to consider Turner syndrome variants in women with unexplained ovarian insufficiency, even in the absence of overt clinical stigmata, to guide appropriate genetic counseling and fertility planning.
期刊介绍:
International Medical Case Reports Journal is an international, peer-reviewed, open access, online journal publishing original case reports from all medical specialties. Submissions should not normally exceed 3,000 words or 4 published pages including figures, diagrams and references. As of 1st April 2019, the International Medical Case Reports Journal will no longer consider meta-analyses for publication.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


