HCNS:A deep learning model for identifying essential proteins based on hypergraph convolution and sequence features
IF 2.5
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
Accurate identification of essential proteins is crucial in biomedical research. Traditional methods rely on Protein–Protein Interaction (PPI) networks and limited biological feature data, often neglecting the critical role of protein amino acid sequences. To address this, we propose the HCNS model, which integrates the Hypergraph Convolutional Network (HGCN) module, the Seq-CNN-MB-NAG feature extraction module, and the Multi-Layer Perceptron (MLP) recognition module, significantly enhancing the accuracy of essential protein identification. The HGCN module constructs a hypergraph from the PPI network and protein complex data, capturing the complex structural relationships between proteins. The Seq-CNN-MB-NAG module utilizes Convolutional Neural Networks (CNN), Multi-Head Self-Attention (MHSA), Bidirectional Long Short-Term Memory (Bi-LSTM), and NAG Transformer to process protein amino acid sequences and extract sequence features. The MLP module then fuses these two sets of features for precise recognition. Experimental results show that the HCNS model outperforms existing methods, achieving an accuracy of 93.38%, with an Area Under the Curve (AUC) of 98.33% and an Area Under the Precision–Recall Curve (AUPR) of 97.16%, demonstrating its potential in essential protein identification.
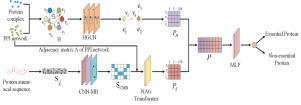
HCNS:一种基于超图卷积和序列特征识别必需蛋白质的深度学习模型
准确鉴定必需蛋白质在生物医学研究中至关重要。传统的方法依赖于蛋白质-蛋白质相互作用(PPI)网络和有限的生物学特征数据,往往忽略了蛋白质氨基酸序列的关键作用。为了解决这个问题,我们提出了HCNS模型,该模型集成了超图卷积网络(HGCN)模块、Seq-CNN-MB-NAG特征提取模块和多层感知器(MLP)识别模块,显著提高了必需蛋白质识别的准确性。HGCN模块利用PPI网络和蛋白质复合体数据构建超图,捕捉蛋白质之间复杂的结构关系。Seq-CNN-MB-NAG模块利用卷积神经网络(CNN)、多头自注意(MHSA)、双向长短期记忆(Bi-LSTM)和NAG Transformer处理蛋白质氨基酸序列并提取序列特征。然后,MLP模块融合这两组特征以进行精确识别。实验结果表明,HCNS模型的准确率为93.38%,曲线下面积(Area Under the Curve, AUC)为98.33%,精确召回曲线下面积(Area Under Precision-Recall Curve, AUPR)为97.16%,优于现有的方法,显示了其在必需蛋白鉴定中的潜力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Analytical biochemistry
生物-分析化学
CiteScore
5.70
自引率
0.00%
发文量
283
审稿时长
44 days
期刊介绍:
The journal''s title Analytical Biochemistry: Methods in the Biological Sciences declares its broad scope: methods for the basic biological sciences that include biochemistry, molecular genetics, cell biology, proteomics, immunology, bioinformatics and wherever the frontiers of research take the field.
The emphasis is on methods from the strictly analytical to the more preparative that would include novel approaches to protein purification as well as improvements in cell and organ culture. The actual techniques are equally inclusive ranging from aptamers to zymology.
The journal has been particularly active in:
-Analytical techniques for biological molecules-
Aptamer selection and utilization-
Biosensors-
Chromatography-
Cloning, sequencing and mutagenesis-
Electrochemical methods-
Electrophoresis-
Enzyme characterization methods-
Immunological approaches-
Mass spectrometry of proteins and nucleic acids-
Metabolomics-
Nano level techniques-
Optical spectroscopy in all its forms.
The journal is reluctant to include most drug and strictly clinical studies as there are more suitable publication platforms for these types of papers.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


