{"title":"Fragment-Based Screening Identifies Novel Non-Amino Acid Inhibitors of the Sodium-Coupled Neutral Amino Acid Transporter SNAT2.","authors":"Sebastian Jakobsen, Carsten Uhd Nielsen","doi":"10.1007/s11095-025-03902-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Amino acid transporters like the sodium-coupled neutral amino acid transporter 2 (SNAT2, SLC38A2) have gained interest for their roles in, e.g., the central nervous system and in cancer. Efforts in discovering inhibitors against these transporters often result in amino acid-based inhibitors that lack selectivity and are likely to compete with amino acid substrates to bind their targets. To circumvent this, we aimed to discover novel non-amino acid inhibitors of SNAT2 by screening a library of fragment-sized compounds.</p><p><strong>Methods: </strong>320 fragment compounds were screened for their inhibition of <sup>3</sup>H-Gly uptake in hyperosmotically upregulated SNAT2-expressing PC-3 cells. The top five hits were studied further for their inhibitory potency and structure-activity relationship (SAR). Their ability to be translocated by SNAT2 was studied using the FLIPR membrane potential (FMP) assay, as well as their mechanism of inhibition.</p><p><strong>Results: </strong>The screen revealed two similar scaffolds that showed SNAT2 inhibition, namely 1,3-benzothiazole-2-amine and 1,3-benzoxazole-2-amine. The SAR revealed how hydrophobic substituents at specific positions were needed for the structures to show SNAT2 inhibition. The best inhibitors inhibited SNAT2 with IC<sub>50</sub>s in the range of 0.64-1.08 mM. Many of the fragment compounds showed an apparent hyperpolarization in the FMP assay, making it difficult to determine their ability to be translocated by SNAT2. An allosteric mechanism of inhibition was implied for the thiazole and oxazole scaffolds, as these resulted in inhibition patterns that resembled non- or un-competitive inhibitors.</p><p><strong>Conclusion: </strong>In conclusion, we discovered multiple novel non-amino acid compounds that inhibited SNAT2 and can serve as starting points for the further development of SNAT2 inhibitors.</p>","PeriodicalId":20027,"journal":{"name":"Pharmaceutical Research","volume":" ","pages":"1285-1297"},"PeriodicalIF":4.3000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12405023/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pharmaceutical Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11095-025-03902-7","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/8 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Amino acid transporters like the sodium-coupled neutral amino acid transporter 2 (SNAT2, SLC38A2) have gained interest for their roles in, e.g., the central nervous system and in cancer. Efforts in discovering inhibitors against these transporters often result in amino acid-based inhibitors that lack selectivity and are likely to compete with amino acid substrates to bind their targets. To circumvent this, we aimed to discover novel non-amino acid inhibitors of SNAT2 by screening a library of fragment-sized compounds.
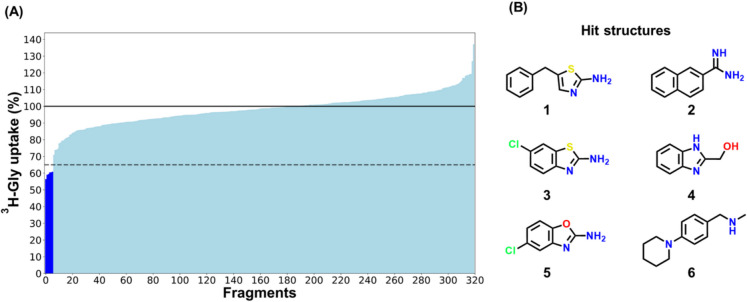
Methods: 320 fragment compounds were screened for their inhibition of 3H-Gly uptake in hyperosmotically upregulated SNAT2-expressing PC-3 cells. The top five hits were studied further for their inhibitory potency and structure-activity relationship (SAR). Their ability to be translocated by SNAT2 was studied using the FLIPR membrane potential (FMP) assay, as well as their mechanism of inhibition.
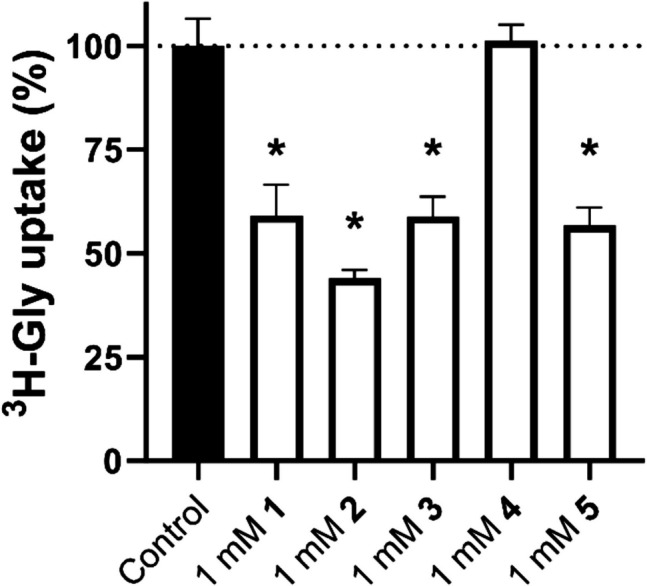
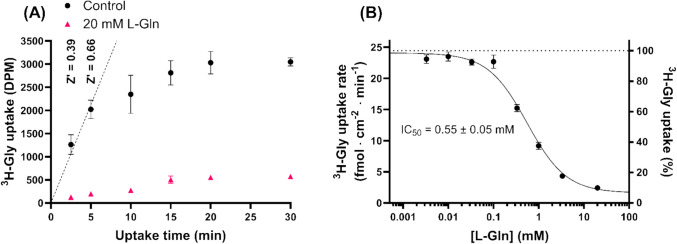
Results: The screen revealed two similar scaffolds that showed SNAT2 inhibition, namely 1,3-benzothiazole-2-amine and 1,3-benzoxazole-2-amine. The SAR revealed how hydrophobic substituents at specific positions were needed for the structures to show SNAT2 inhibition. The best inhibitors inhibited SNAT2 with IC50s in the range of 0.64-1.08 mM. Many of the fragment compounds showed an apparent hyperpolarization in the FMP assay, making it difficult to determine their ability to be translocated by SNAT2. An allosteric mechanism of inhibition was implied for the thiazole and oxazole scaffolds, as these resulted in inhibition patterns that resembled non- or un-competitive inhibitors.
Conclusion: In conclusion, we discovered multiple novel non-amino acid compounds that inhibited SNAT2 and can serve as starting points for the further development of SNAT2 inhibitors.
期刊介绍:
Pharmaceutical Research, an official journal of the American Association of Pharmaceutical Scientists, is committed to publishing novel research that is mechanism-based, hypothesis-driven and addresses significant issues in drug discovery, development and regulation. Current areas of interest include, but are not limited to:
-(pre)formulation engineering and processing-
computational biopharmaceutics-
drug delivery and targeting-
molecular biopharmaceutics and drug disposition (including cellular and molecular pharmacology)-
pharmacokinetics, pharmacodynamics and pharmacogenetics.
Research may involve nonclinical and clinical studies, and utilize both in vitro and in vivo approaches. Studies on small drug molecules, pharmaceutical solid materials (including biomaterials, polymers and nanoparticles) biotechnology products (including genes, peptides, proteins and vaccines), and genetically engineered cells are welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


