Jana Horackova, Renata Taslerova, Milan Bayer, Jana Nekvindova, Ladislava Pavlikova, Jan M Horacek, Vladimir Palicka
{"title":"Juvenile Paget disease with unique compound heterozygous sequence variants in the TNFRSF11B gene.","authors":"Jana Horackova, Renata Taslerova, Milan Bayer, Jana Nekvindova, Ladislava Pavlikova, Jan M Horacek, Vladimir Palicka","doi":"10.1186/s13023-025-03804-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Juvenile Paget disease (JPD) is a rare autosomal recessive bone disease characterized by escalated bone metabolism leading to skeletal deformities, susceptibility to fractures, and some extraskeletal findings. This genetic disease is associated with changes in the TNFRSF11B gene encoding osteoprotegerin, an important regulator of osteoresorption. Most published JPD cases have been found to carry homozygous TNFRSF11B variants, while compound heterozygous variants in this gene have been reported only twice.</p><p><strong>Methods and results: </strong>We report the first case of JPD diagnosed in the Czech Republic, who presented with a mild phenotype of this disease. The first bone fractures, appeared at 3 years of age. Other clinical manifestations included typical skeletal deformities, macrocephaly, arched chest, lower extremity valgosity, lateral bowing of the thighs, and anterior bowing of the shins. Minor mixed hearing impairment, angioid stripes of the choroidea, and temporary immunodeficiency were present among extra-skeletal findings. Sanger sequencing was performed on both the patient and the parents to test for the presence of TNFRSF11B sequence variants. Molecular genetic analysis showed unique compound heterozygous sequence variants in TNFRSF11B: a paternally inherited variant c.30 + 5G > A, p.(?) and a maternally inherited variant c.329G > T, p.(Gly110Val). Both of the variants were analyzed by several in silico predictive tools indicating, for their strongly supported pathogenicity according to American College of Medical Genetics and Genomics standards. Furthermore, we present diagnostic findings, their treatment, and follow-up care.</p><p><strong>Conclusion: </strong>The newly described variants of TNFRSF11B extend knowledge of this very rare disease. Early diagnosis and antiresorption treatment prevent further fractures and deformity progression, and improve the patient's quality of life. This example of osteoprotegerin deficiency may help us better understand its role in skeletal and non-skeletal systems.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"409"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12333066/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03804-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Juvenile Paget disease (JPD) is a rare autosomal recessive bone disease characterized by escalated bone metabolism leading to skeletal deformities, susceptibility to fractures, and some extraskeletal findings. This genetic disease is associated with changes in the TNFRSF11B gene encoding osteoprotegerin, an important regulator of osteoresorption. Most published JPD cases have been found to carry homozygous TNFRSF11B variants, while compound heterozygous variants in this gene have been reported only twice.
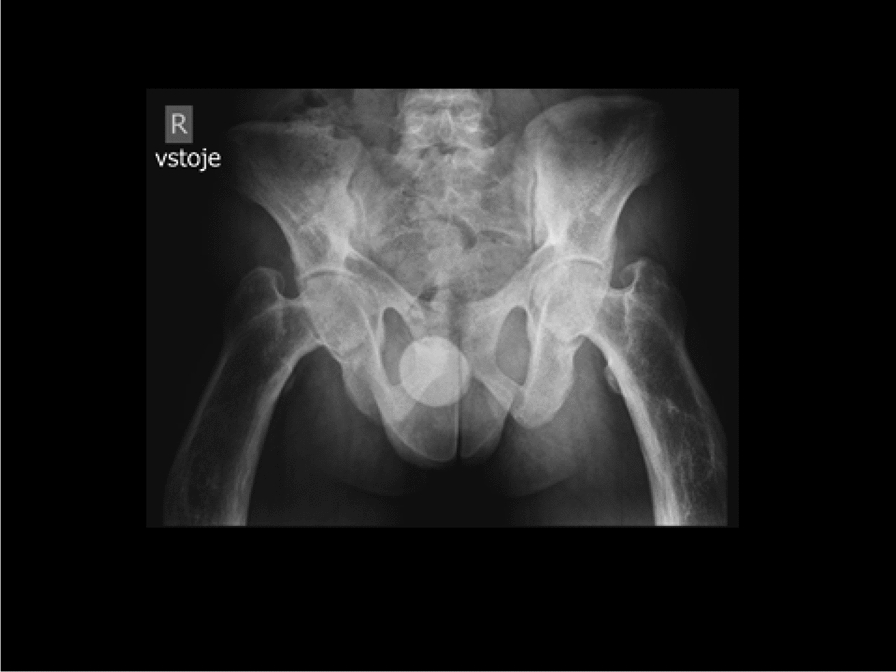
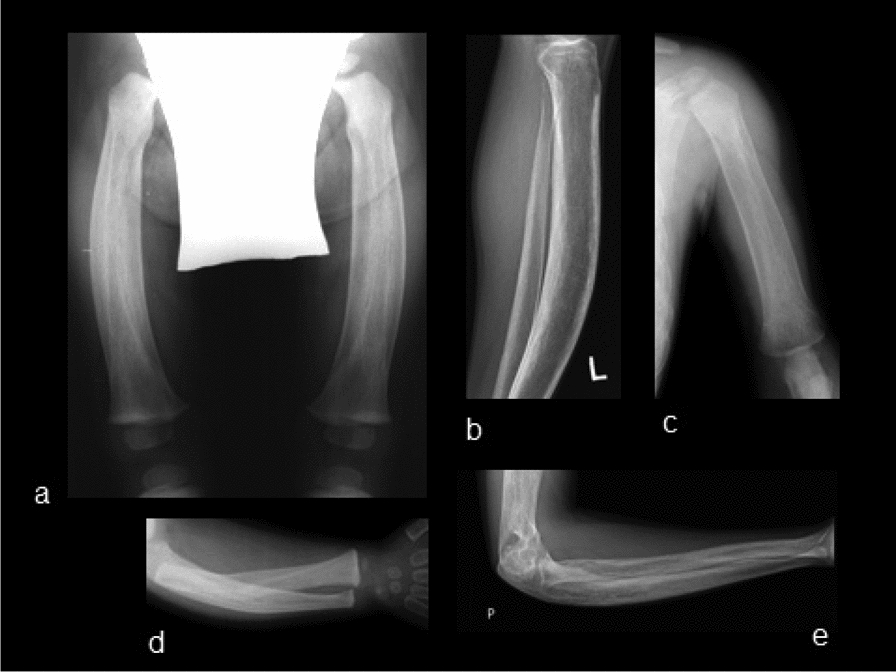
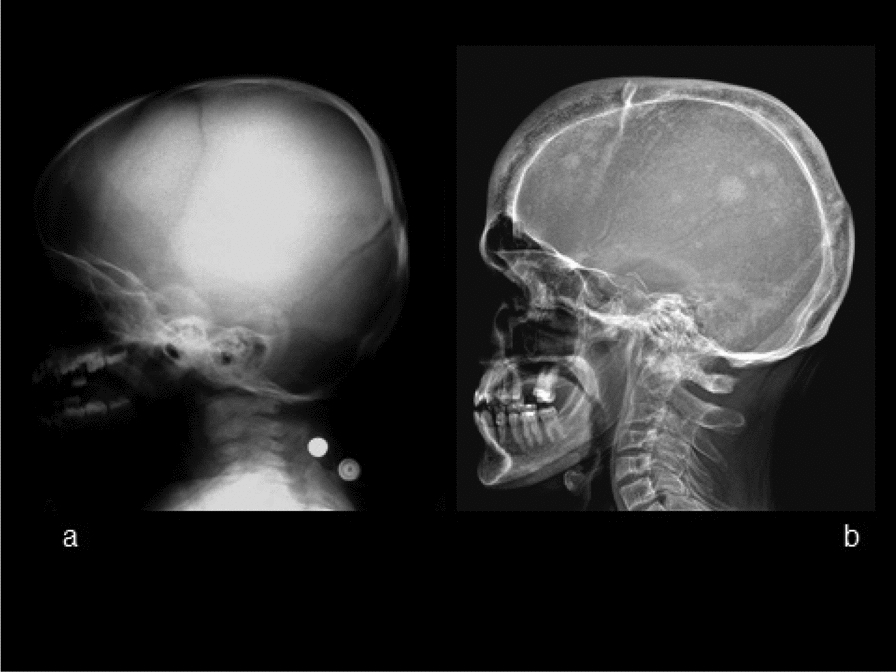
Methods and results: We report the first case of JPD diagnosed in the Czech Republic, who presented with a mild phenotype of this disease. The first bone fractures, appeared at 3 years of age. Other clinical manifestations included typical skeletal deformities, macrocephaly, arched chest, lower extremity valgosity, lateral bowing of the thighs, and anterior bowing of the shins. Minor mixed hearing impairment, angioid stripes of the choroidea, and temporary immunodeficiency were present among extra-skeletal findings. Sanger sequencing was performed on both the patient and the parents to test for the presence of TNFRSF11B sequence variants. Molecular genetic analysis showed unique compound heterozygous sequence variants in TNFRSF11B: a paternally inherited variant c.30 + 5G > A, p.(?) and a maternally inherited variant c.329G > T, p.(Gly110Val). Both of the variants were analyzed by several in silico predictive tools indicating, for their strongly supported pathogenicity according to American College of Medical Genetics and Genomics standards. Furthermore, we present diagnostic findings, their treatment, and follow-up care.
Conclusion: The newly described variants of TNFRSF11B extend knowledge of this very rare disease. Early diagnosis and antiresorption treatment prevent further fractures and deformity progression, and improve the patient's quality of life. This example of osteoprotegerin deficiency may help us better understand its role in skeletal and non-skeletal systems.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


