The Platinum Pedigree: a long-read benchmark for genetic variants
IF 32.1
1区 生物学
Q1 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
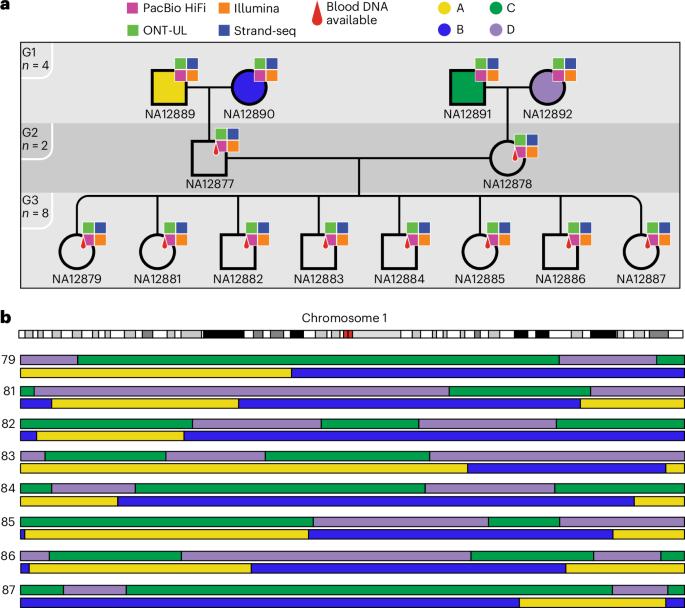
Recent advances in genome sequencing have improved variant calling in complex regions of the human genome. However, it is difficult to quantify variant calling performance because existing standards often focus on specificity, neglecting completeness in difficult-to-analyze regions. To create a more comprehensive truth set, we used Mendelian inheritance in a large pedigree (CEPH-1463) to filter variants across PacBio high-fidelity (HiFi), Illumina and Oxford Nanopore Technologies platforms. This generated a variant map with over 4.7 million single-nucleotide variants, 767,795 insertions and deletions (indels), 537,486 tandem repeats and 24,315 structural variants, covering 2.77 Gb of the GRCh38 genome. This work adds ~200 Mb of high-confidence regions, including 8% more small variants, and introduces the first tandem repeat and structural variant truth sets for NA12878 and her family. As an example of the value of this improved benchmark, we retrained DeepVariant using these data to reduce genotyping errors by ~34%. This work introduces a pedigree-derived benchmark for single-nucleotide variants, indels, structural variants and tandem repeats, offering a variant map to validate sequencing workflows or to support the development and evaluation of new variant callers.
白金谱系:一个长期阅读的基因变异基准。
基因组测序的最新进展改善了人类基因组复杂区域的变异召唤。然而,由于现有的标准往往侧重于特异性,而忽略了难以分析区域的完整性,因此很难对变量调用性能进行量化。为了创建一个更全面的真相集,我们在一个大谱系(CEPH-1463)中使用孟德尔遗传来过滤PacBio高保真(HiFi)、Illumina和Oxford Nanopore Technologies平台上的变异。该变异图谱包含470多万个单核苷酸变异、767,795个插入和缺失(索引)、537,486个串联重复序列和24,315个结构变异,覆盖了2.77 Gb的GRCh38基因组。这项工作增加了约200mb的高置信度区域,其中包括8%的小变异,并为NA12878及其家族引入了第一个串联重复和结构变异真值集。作为这个改进的基准价值的一个例子,我们使用这些数据重新训练DeepVariant,将基因分型错误减少了约34%。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Methods
生物-生化研究方法
CiteScore
58.70
自引率
1.70%
发文量
326
审稿时长
1 months
期刊介绍:
Nature Methods is a monthly journal that focuses on publishing innovative methods and substantial enhancements to fundamental life sciences research techniques. Geared towards a diverse, interdisciplinary readership of researchers in academia and industry engaged in laboratory work, the journal offers new tools for research and emphasizes the immediate practical significance of the featured work. It publishes primary research papers and reviews recent technical and methodological advancements, with a particular interest in primary methods papers relevant to the biological and biomedical sciences. This includes methods rooted in chemistry with practical applications for studying biological problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


