Practical Machine Learning Strategies 4: Using Neural Networks to Replicate Proton and 13C NMR Chemical Shifts Obtained from ωB97X-D/6-31G* Density Functional Calculations
Thomas Hehre, Philip E. Klunzinger, Bernard J. Deppmeier, William Sean Ohlinger and Warren J Hehre*,
{"title":"Practical Machine Learning Strategies 4: Using Neural Networks to Replicate Proton and 13C NMR Chemical Shifts Obtained from ωB97X-D/6-31G* Density Functional Calculations","authors":"Thomas Hehre, Philip E. Klunzinger, Bernard J. Deppmeier, William Sean Ohlinger and Warren J Hehre*, ","doi":"10.1021/acs.joc.5c00927","DOIUrl":null,"url":null,"abstract":"<p >Described are neural networks that accurately reproduce proton and <sup>13</sup>C chemical shifts obtained from ωB97X-D/6-31G*//ωB97X-D/6-31G* density functional model GIAO calculations. They support uncharged, closed-shell molecules comprising H, C, N, O, F, S, Cl, and Br. Development involved training to ≈2.7 million equilibrium geometry and chemical shift calculations for a diverse collection of organic molecules (including synthetic drugs and natural products). Referenced to ωB97X-D/6-31G*//ωB97X-D/6-31G* calculations, chemical shifts from neural networks for 601 marine natural products show RMS errors of 0.05 ppm (proton) and 0.76 ppm (<sup>13</sup>C). RMS errors of 0.09 ppm (proton) and 1.02 ppm (<sup>13</sup>C) shifts result when equilibrium geometries from a previously described “estimated ωB97X-D/6-31G*” neural network model (trained to reproduce ωB97X-D/6-31G* geometries) are utilized. A second assessment of experimental <sup>13</sup>C chemical shifts for 246 natural products is provided. Using neural network models to provide both geometries and chemical shifts: 45% of <sup>13</sup>C shifts reproduce experimental values within 1 ppm, 73% within 2 ppm, and 86% within 3 ppm. Utilizing neural network models for both equilibrium geometries and chemical shifts reduces the computational time required for accurate proton and <sup>13</sup>C chemical shifts from tens to hundreds of minutes to just a few seconds per molecule.</p>","PeriodicalId":57,"journal":{"name":"Journal of Organic Chemistry","volume":"90 32","pages":"11478–11485"},"PeriodicalIF":3.6000,"publicationDate":"2025-08-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Organic Chemistry","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.joc.5c00927","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
引用次数: 0
Abstract
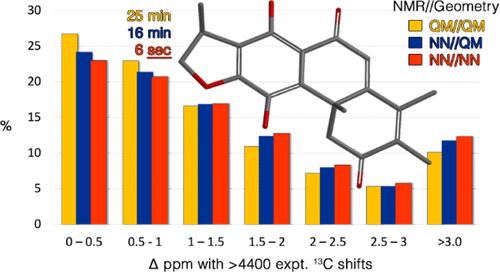
Described are neural networks that accurately reproduce proton and 13C chemical shifts obtained from ωB97X-D/6-31G*//ωB97X-D/6-31G* density functional model GIAO calculations. They support uncharged, closed-shell molecules comprising H, C, N, O, F, S, Cl, and Br. Development involved training to ≈2.7 million equilibrium geometry and chemical shift calculations for a diverse collection of organic molecules (including synthetic drugs and natural products). Referenced to ωB97X-D/6-31G*//ωB97X-D/6-31G* calculations, chemical shifts from neural networks for 601 marine natural products show RMS errors of 0.05 ppm (proton) and 0.76 ppm (13C). RMS errors of 0.09 ppm (proton) and 1.02 ppm (13C) shifts result when equilibrium geometries from a previously described “estimated ωB97X-D/6-31G*” neural network model (trained to reproduce ωB97X-D/6-31G* geometries) are utilized. A second assessment of experimental 13C chemical shifts for 246 natural products is provided. Using neural network models to provide both geometries and chemical shifts: 45% of 13C shifts reproduce experimental values within 1 ppm, 73% within 2 ppm, and 86% within 3 ppm. Utilizing neural network models for both equilibrium geometries and chemical shifts reduces the computational time required for accurate proton and 13C chemical shifts from tens to hundreds of minutes to just a few seconds per molecule.
期刊介绍:
Journal of Organic Chemistry welcomes original contributions of fundamental research in all branches of the theory and practice of organic chemistry. In selecting manuscripts for publication, the editors place emphasis on the quality and novelty of the work, as well as the breadth of interest to the organic chemistry community.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


