Harriet Longley, David Bonsall, Joshua Herbeck, George MacIntyre-Cockett, Sandra E Chaudron, Laura Thomson, Nicholas Grayson, Andrew Mujugira, Christophe Fraser, Jairam Lingappa, Katrina Lythgoe
{"title":"Estimates of HIV-1 within-host recombination rates across the whole genome.","authors":"Harriet Longley, David Bonsall, Joshua Herbeck, George MacIntyre-Cockett, Sandra E Chaudron, Laura Thomson, Nicholas Grayson, Andrew Mujugira, Christophe Fraser, Jairam Lingappa, Katrina Lythgoe","doi":"10.1093/ve/veaf052","DOIUrl":null,"url":null,"abstract":"<p><p>Recombination plays a pivotal role in generating within-host diversity and enabling HIV's evolutionary success, particularly in evading the host immune response. Despite this, the variability in recombination rates across different settings and the underlying factors that drive these differences remain poorly understood. In this study, we analysed a large dataset encompassing hundreds of untreated, longitudinally sampled infections using both whole-genome long-read and short-read sequencing datasets. By quantifying recombination rates, we uncover substantial variation across subtypes, viral loads, and stages of infection. We also map recombination hot and cold spots across the genome using a sliding window approach, finding that previously reported inter-subtype regions of high or low recombination are replicated at the within-host level. Importantly, our findings reveal the significant influence of selection on recombination, showing that the presence and success of recombinant genomes is strongly interconnected with the fitness landscape. These results offer valuable insights into the contribution of recombination to evolutionary dynamics and demonstrate the enhanced resolution that long-read sequencing offers for studying viral evolution.</p>","PeriodicalId":56026,"journal":{"name":"Virus Evolution","volume":"11 1","pages":"veaf052"},"PeriodicalIF":4.0000,"publicationDate":"2025-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12309388/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Virus Evolution","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/ve/veaf052","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"VIROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
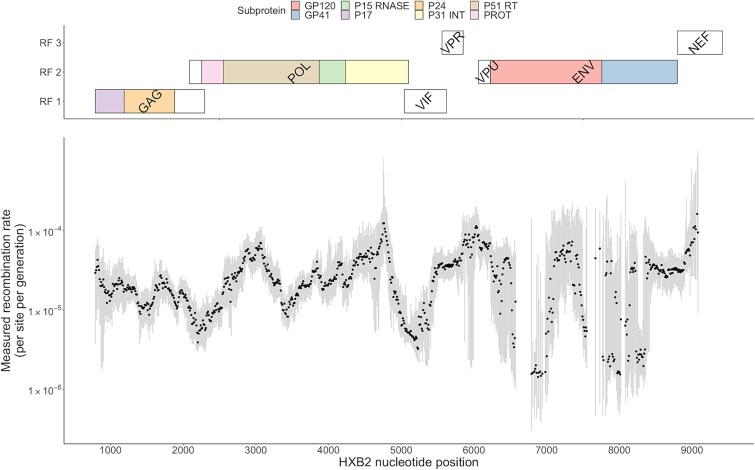
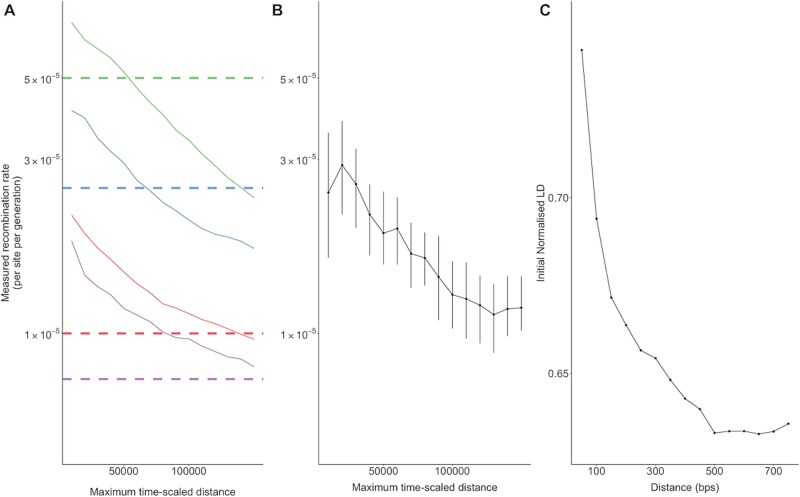
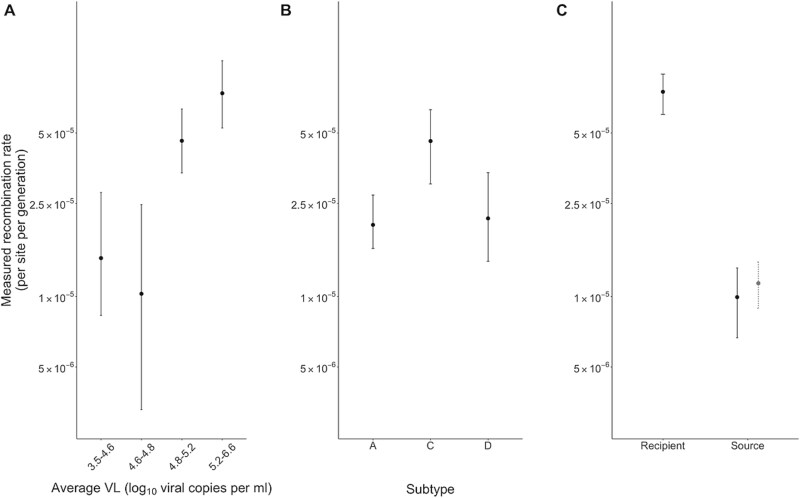
Recombination plays a pivotal role in generating within-host diversity and enabling HIV's evolutionary success, particularly in evading the host immune response. Despite this, the variability in recombination rates across different settings and the underlying factors that drive these differences remain poorly understood. In this study, we analysed a large dataset encompassing hundreds of untreated, longitudinally sampled infections using both whole-genome long-read and short-read sequencing datasets. By quantifying recombination rates, we uncover substantial variation across subtypes, viral loads, and stages of infection. We also map recombination hot and cold spots across the genome using a sliding window approach, finding that previously reported inter-subtype regions of high or low recombination are replicated at the within-host level. Importantly, our findings reveal the significant influence of selection on recombination, showing that the presence and success of recombinant genomes is strongly interconnected with the fitness landscape. These results offer valuable insights into the contribution of recombination to evolutionary dynamics and demonstrate the enhanced resolution that long-read sequencing offers for studying viral evolution.
期刊介绍:
Virus Evolution is a new Open Access journal focusing on the long-term evolution of viruses, viruses as a model system for studying evolutionary processes, viral molecular epidemiology and environmental virology.
The aim of the journal is to provide a forum for original research papers, reviews, commentaries and a venue for in-depth discussion on the topics relevant to virus evolution.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


