Abril Morales, Elena Korsakova, Niloufar Mansooralavi, Peter Soliman, Sarvin Jahanbani, Michelle L Olsen, Aparna Badhuri, William E Lowry
{"title":"Evidence of neuronal DNA damage in the brains of patients with Rett syndrome.","authors":"Abril Morales, Elena Korsakova, Niloufar Mansooralavi, Peter Soliman, Sarvin Jahanbani, Michelle L Olsen, Aparna Badhuri, William E Lowry","doi":"10.1242/dmm.052358","DOIUrl":null,"url":null,"abstract":"<p><p>Rett syndrome is characterized by the postnatal loss of neurophysiological function and regression of childhood development. Because the syndrome is X linked, and males with MECP2 mutations generally do not survive birth, the study of this syndrome has been complicated by the fact that, in the female brain, a portion of neurons express wild-type MECP2, and another portion of neurons express a non-functional allele of MECP2. Here, we present an approach that enables transcriptional profiling of individual neurons and direct comparison of neurons that express functional MECP2 with those that have diminished MECP2 function. With this novel profiling approach, we found that mutant neurons from the brains of patients with Rett syndrome show patterns of defects in expression of synaptic and metabolic genes. A similar analysis of rat brain lacking MECP2 expression yielded similar patterns, suggesting that rat is a suitable in vivo model of Rett syndrome. These analyses also identified DNA damage and senescence transcriptional signatures specifically in MECP2-null neurons, suggesting a possible trigger of dysfunction in Rett syndrome. Together, these data highlight potentially defective molecular, physiological and metabolic pathways in brain neurons of patients with Rett syndrome.</p>","PeriodicalId":11144,"journal":{"name":"Disease Models & Mechanisms","volume":" ","pages":""},"PeriodicalIF":3.3000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12452059/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Disease Models & Mechanisms","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1242/dmm.052358","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/9/1 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
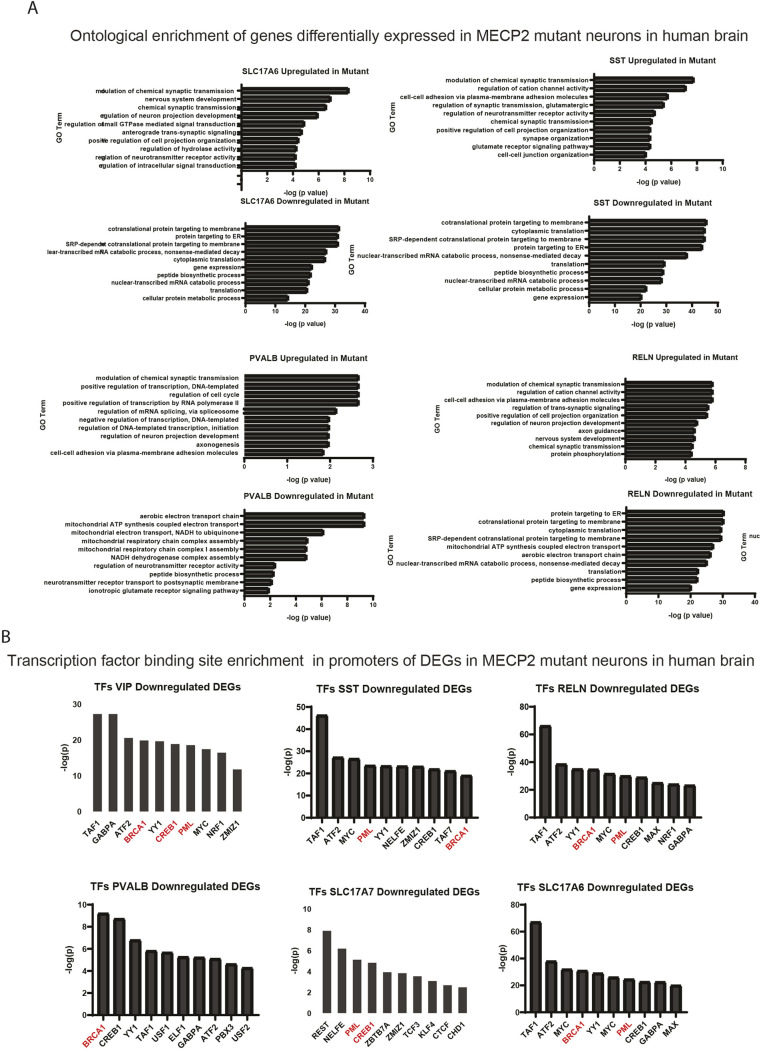
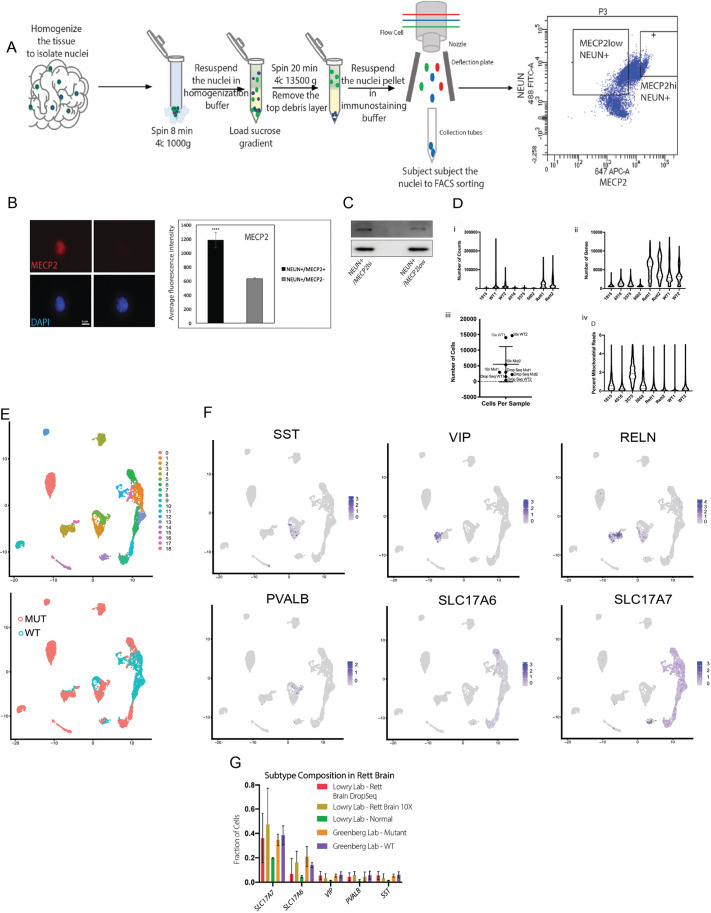
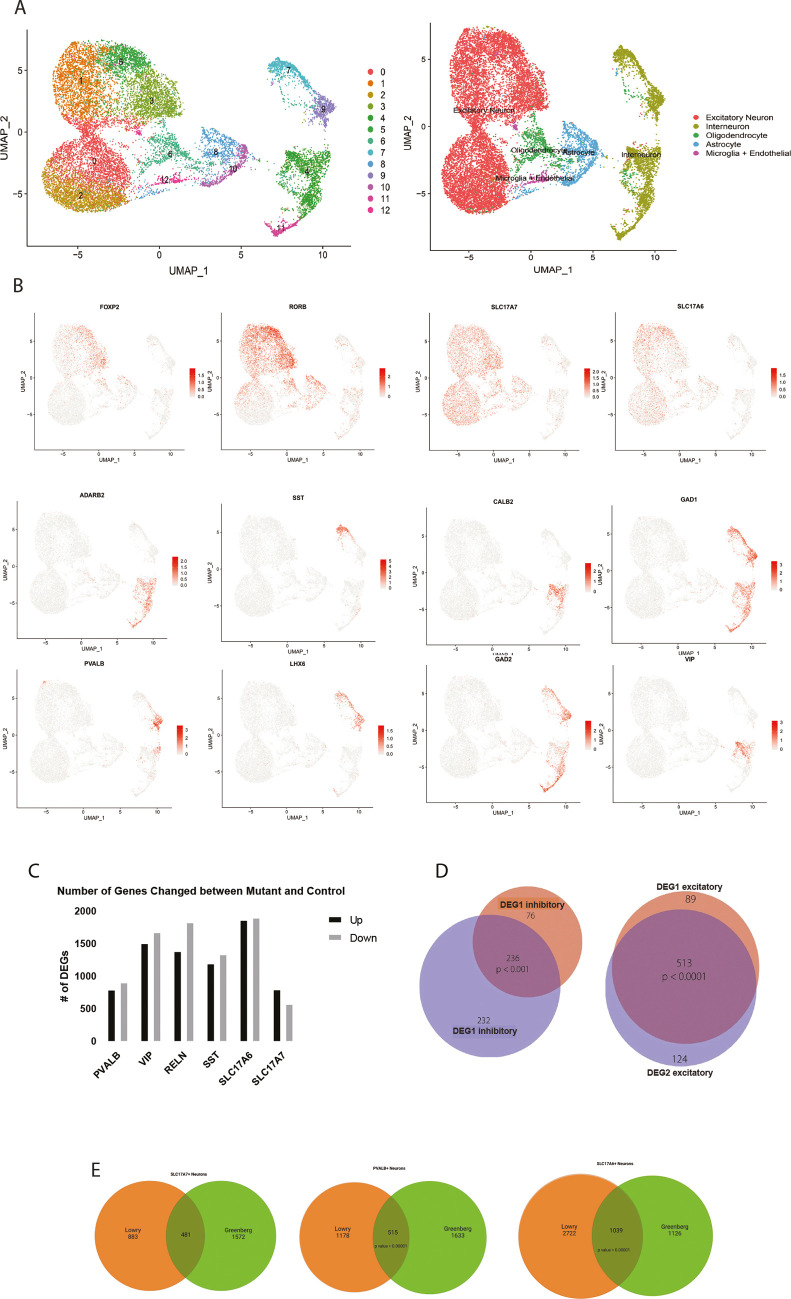
Rett syndrome is characterized by the postnatal loss of neurophysiological function and regression of childhood development. Because the syndrome is X linked, and males with MECP2 mutations generally do not survive birth, the study of this syndrome has been complicated by the fact that, in the female brain, a portion of neurons express wild-type MECP2, and another portion of neurons express a non-functional allele of MECP2. Here, we present an approach that enables transcriptional profiling of individual neurons and direct comparison of neurons that express functional MECP2 with those that have diminished MECP2 function. With this novel profiling approach, we found that mutant neurons from the brains of patients with Rett syndrome show patterns of defects in expression of synaptic and metabolic genes. A similar analysis of rat brain lacking MECP2 expression yielded similar patterns, suggesting that rat is a suitable in vivo model of Rett syndrome. These analyses also identified DNA damage and senescence transcriptional signatures specifically in MECP2-null neurons, suggesting a possible trigger of dysfunction in Rett syndrome. Together, these data highlight potentially defective molecular, physiological and metabolic pathways in brain neurons of patients with Rett syndrome.
期刊介绍:
Disease Models & Mechanisms (DMM) is an online Open Access journal focusing on the use of model systems to better understand, diagnose and treat human disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


