{"title":"Mevalonate kinase deficiency: genetic and clinical characteristics of a Chinese pediatric cohort.","authors":"Chenchen Guan, Wenjie Wang, Qinhua Zhou, Jinqiao Sun, Lipin Liu, Luyao Liu, Bijun Sun, Jia Hou, Xiaochuan Wang","doi":"10.1186/s12969-025-01131-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Mevalonate kinase deficiency (MKD) is a rare autoinflammatory disease, and mevalonic aciduria (MA) is a severe phenotype of MKD. The present study reports the characteristics of MKD and four novel mutations in the mevalonate kinase (MVK) gene in a Chinese cohort.</p><p><strong>Method: </strong>A retrospective study was conducted on patients diagnosed with MKD from July 2013 to December 2024. The clinical, immunological, and follow-up data were collected from electronic medical records. Next-generation sequencing and Sanger sequencing were performed to identify gene mutations. A literature review was performed on MKD patients to further investigate the associations between genotype and phenotype.</p><p><strong>Results: </strong>Eleven MKD patients were enrolled from a Chinese cohort of prolonged and recurrent fever of unknown origin. Ten patients were classified as having hyperimmunoglobulin D syndrome (HIDS), and one patient was classified as having MA. The median follow-up duration was 5 years (IQR: 1.5-6 years). Recurrent fever and gastrointestinal symptoms were the most common symptoms. Anemia was observed in 8 of the 11 patients, including one patient with severe hematological complications. Growth restriction (5/11 patients) and developmental delay (4/11 patients) were also observed. IgD levels were measured in ten patients, and the median IgD level was 85.23 µg/ml (IQR: 18.74-385.19 µg/ml). Four novel mutation sites in the MVK gene were discovered: c.78G > A, c.463G > A, c.1076C > T and c.1088G > A. Etanercept was the effective biological agent tested, leading to complete or partial remission in 5 of the 6 patients. A literature review of 20 MA patients suggested that homozygous MVK gene mutations are more frequently associated with MA. Moreover, MA patients with the homozygous A334T mutation present a milder phenotype, and those with the I268T homozygous mutation present a more severe phenotype.</p><p><strong>Conclusions: </strong>This study is the largest case series of MKD pediatric patients from China. Four new mutation sites in MVK were identified, further expanding the phenotypic and genotypic spectrum of MKD and emphasizing the significance of MVK mutation patterns in influencing disease severity.</p>","PeriodicalId":54630,"journal":{"name":"Pediatric Rheumatology","volume":"23 1","pages":"78"},"PeriodicalIF":2.3000,"publicationDate":"2025-07-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12297814/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pediatric Rheumatology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12969-025-01131-1","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Mevalonate kinase deficiency (MKD) is a rare autoinflammatory disease, and mevalonic aciduria (MA) is a severe phenotype of MKD. The present study reports the characteristics of MKD and four novel mutations in the mevalonate kinase (MVK) gene in a Chinese cohort.
Method: A retrospective study was conducted on patients diagnosed with MKD from July 2013 to December 2024. The clinical, immunological, and follow-up data were collected from electronic medical records. Next-generation sequencing and Sanger sequencing were performed to identify gene mutations. A literature review was performed on MKD patients to further investigate the associations between genotype and phenotype.
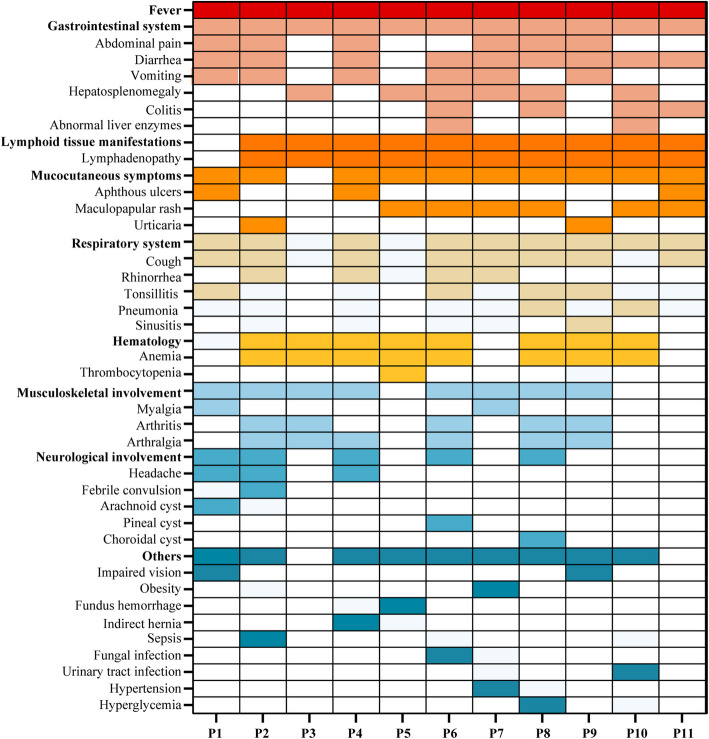
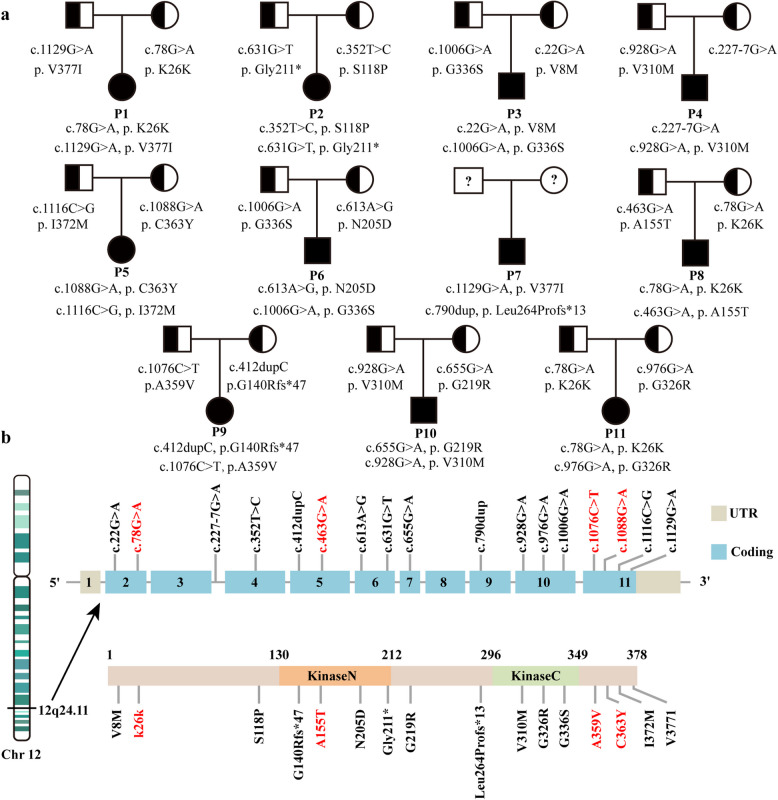
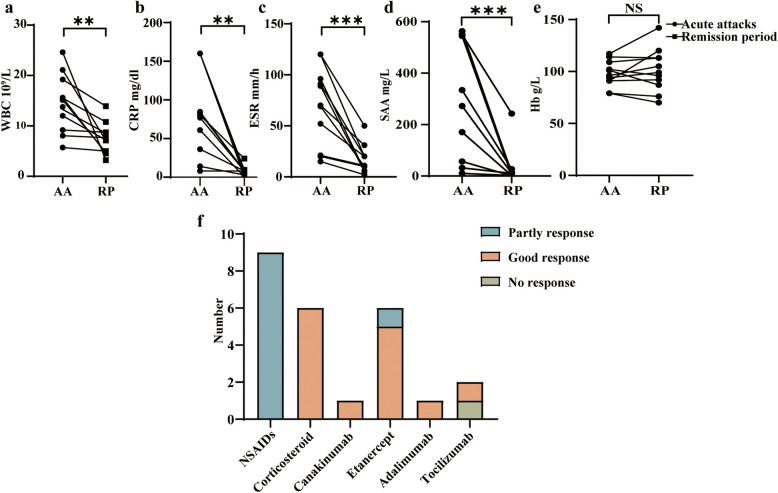
Results: Eleven MKD patients were enrolled from a Chinese cohort of prolonged and recurrent fever of unknown origin. Ten patients were classified as having hyperimmunoglobulin D syndrome (HIDS), and one patient was classified as having MA. The median follow-up duration was 5 years (IQR: 1.5-6 years). Recurrent fever and gastrointestinal symptoms were the most common symptoms. Anemia was observed in 8 of the 11 patients, including one patient with severe hematological complications. Growth restriction (5/11 patients) and developmental delay (4/11 patients) were also observed. IgD levels were measured in ten patients, and the median IgD level was 85.23 µg/ml (IQR: 18.74-385.19 µg/ml). Four novel mutation sites in the MVK gene were discovered: c.78G > A, c.463G > A, c.1076C > T and c.1088G > A. Etanercept was the effective biological agent tested, leading to complete or partial remission in 5 of the 6 patients. A literature review of 20 MA patients suggested that homozygous MVK gene mutations are more frequently associated with MA. Moreover, MA patients with the homozygous A334T mutation present a milder phenotype, and those with the I268T homozygous mutation present a more severe phenotype.
Conclusions: This study is the largest case series of MKD pediatric patients from China. Four new mutation sites in MVK were identified, further expanding the phenotypic and genotypic spectrum of MKD and emphasizing the significance of MVK mutation patterns in influencing disease severity.
期刊介绍:
Pediatric Rheumatology is an open access, peer-reviewed, online journal encompassing all aspects of clinical and basic research related to pediatric rheumatology and allied subjects.
The journal’s scope of diseases and syndromes include musculoskeletal pain syndromes, rheumatic fever and post-streptococcal syndromes, juvenile idiopathic arthritis, systemic lupus erythematosus, juvenile dermatomyositis, local and systemic scleroderma, Kawasaki disease, Henoch-Schonlein purpura and other vasculitides, sarcoidosis, inherited musculoskeletal syndromes, autoinflammatory syndromes, and others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


