Erta Rajabi, Mahsa Choroom Kheirabadi, Nasrin Alipour Olyaei, Anne Molitor, Mohadese Sadat Mousavi Khorshidi, Morteza Heidari, Arash Abbasi, Parastoo Rostami, Mohadese Mahdavi, Raphael Carapito, Mohammad Shahrooei, Seiamak Bahram, Nima Parvaneh
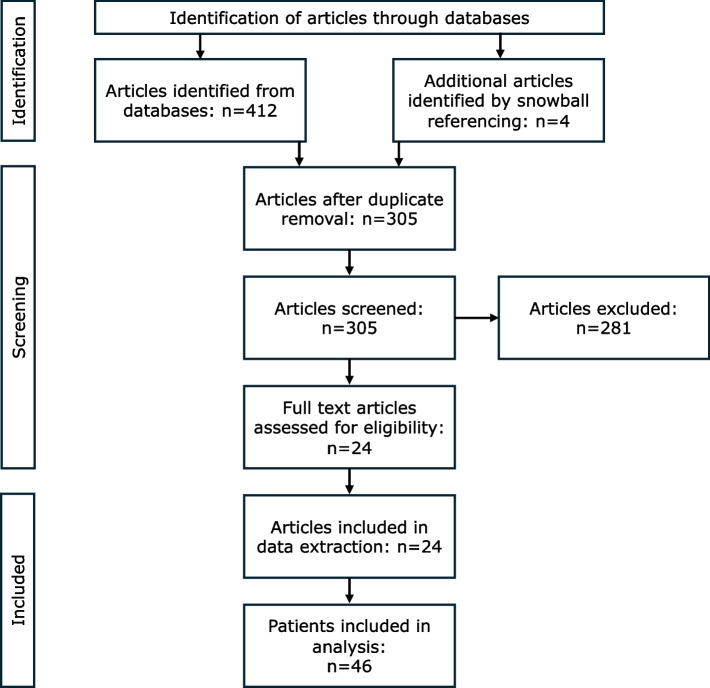
{"title":"Complete Complement Factor I (CFI) deficiency: a systematic review of forty-nine patients including three novel cases.","authors":"Erta Rajabi, Mahsa Choroom Kheirabadi, Nasrin Alipour Olyaei, Anne Molitor, Mohadese Sadat Mousavi Khorshidi, Morteza Heidari, Arash Abbasi, Parastoo Rostami, Mohadese Mahdavi, Raphael Carapito, Mohammad Shahrooei, Seiamak Bahram, Nima Parvaneh","doi":"10.1186/s12865-025-00739-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Complete complement factor I (CFI) deficiency is an inborn error of immunity (IEI) that results in heightened susceptibility to infections and immune dysregulatory disorders. This systematic review seeks to enhance our understanding of the clinical characteristics, genotype-phenotype correlations, and treatment outcomes in patients with complete CFI deficiency, including three novel cases. We conducted a comprehensive literature review of cases published from 1996 to November 2024, identifying 49 patients with homozygous or compound heterozygous mutations in the CFI gene.</p><p><strong>Results: </strong>Among the 49 patients, the mean age at initial presentation was 7.19 (± SD: 9.75) years. Most patients presented with infectious manifestations (n: 37, 75.5%), particularly sepsis (n: 18, 36.7%). The predominant pathogens were encapsulated organisms, particularly Neisseria meningitidis. Immune dysregulatory manifestations involved rheumatologic (n: 14, 28.57%), neurologic (n: 11, 22.4%), and renal (n: 8, 16.3%) disorders. Immunological evaluations showed low or absent levels of C3 and CFI in most patients. Genetic analysis identified 45 distinct mutations; less deleterious mutations, such as missense and splicing variants, were more common in those with immune dysregulation. Notably, three patients treated with eculizumab demonstrated significant clinical improvement.</p><p><strong>Conclusion: </strong>Complete CFI deficiency presents a varied clinical spectrum, from asymptomatic to recurrent infections and immune dysregulation. Early diagnosis and targeted therapies, such as eculizumab, may improve patient outcomes. These findings underscore the necessity for further research into the nature of complete CFI deficiency and the development of optimal management strategies.</p>","PeriodicalId":9040,"journal":{"name":"BMC Immunology","volume":"26 1","pages":"54"},"PeriodicalIF":2.7000,"publicationDate":"2025-07-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12297857/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12865-025-00739-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Complete complement factor I (CFI) deficiency is an inborn error of immunity (IEI) that results in heightened susceptibility to infections and immune dysregulatory disorders. This systematic review seeks to enhance our understanding of the clinical characteristics, genotype-phenotype correlations, and treatment outcomes in patients with complete CFI deficiency, including three novel cases. We conducted a comprehensive literature review of cases published from 1996 to November 2024, identifying 49 patients with homozygous or compound heterozygous mutations in the CFI gene.
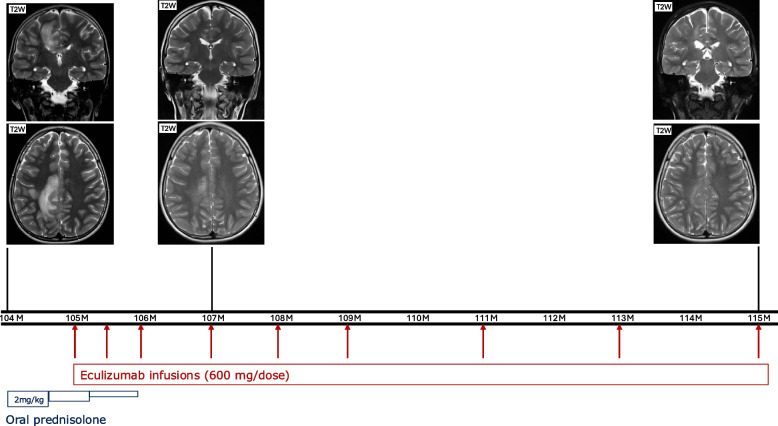
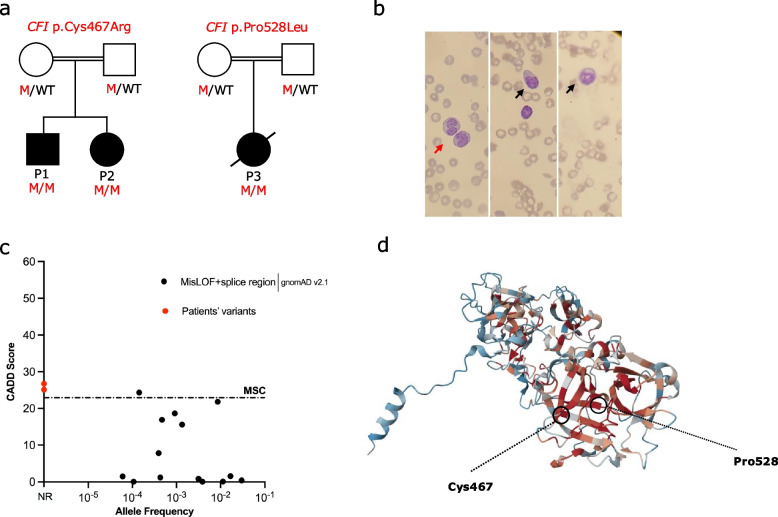
Results: Among the 49 patients, the mean age at initial presentation was 7.19 (± SD: 9.75) years. Most patients presented with infectious manifestations (n: 37, 75.5%), particularly sepsis (n: 18, 36.7%). The predominant pathogens were encapsulated organisms, particularly Neisseria meningitidis. Immune dysregulatory manifestations involved rheumatologic (n: 14, 28.57%), neurologic (n: 11, 22.4%), and renal (n: 8, 16.3%) disorders. Immunological evaluations showed low or absent levels of C3 and CFI in most patients. Genetic analysis identified 45 distinct mutations; less deleterious mutations, such as missense and splicing variants, were more common in those with immune dysregulation. Notably, three patients treated with eculizumab demonstrated significant clinical improvement.
Conclusion: Complete CFI deficiency presents a varied clinical spectrum, from asymptomatic to recurrent infections and immune dysregulation. Early diagnosis and targeted therapies, such as eculizumab, may improve patient outcomes. These findings underscore the necessity for further research into the nature of complete CFI deficiency and the development of optimal management strategies.
期刊介绍:
BMC Immunology is an open access journal publishing original peer-reviewed research articles in molecular, cellular, tissue-level, organismal, functional, and developmental aspects of the immune system as well as clinical studies and animal models of human diseases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


