Automated identification of small molecules in cryoelectron microscopy data with density- and energy-guided evaluation
IF 4.3
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
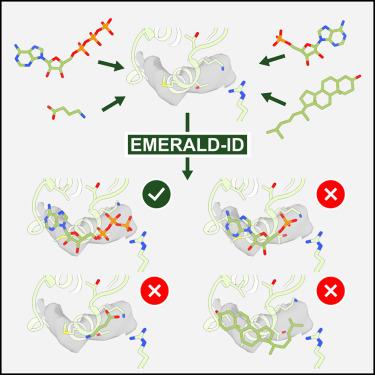
Methodological improvements in cryoelectron microscopy (cryo-EM) have made it useful in ligand-bound structure determination for biology and drug design. However, determining ligand conformation and identity is challenging at the resolutions typical for cryo-EM. Automated methods can aid in ligand conformational modeling, but current ligand identification tools—developed for X-ray crystallography data—perform poorly at resolutions common for cryo-EM. Here, we present EMERALD-ID, a method capable of docking and evaluating small molecule conformations for ligand identification. EMERALD-ID identifies 44% of common ligands exactly and identifies closely related ligands in 66% of cases. We then use this tool to discover possible ligand identification errors, as well as previously unidentified ligands. Furthermore, we show EMERALD-ID identifying ligands from custom ligand libraries of various small molecule types, including human metabolites and drug fragments. Our method provides a valuable addition to cryo-EM modeling tools to improve small molecule model accuracy and quality.
用密度和能量引导评价的低温电子显微镜数据中的小分子的自动鉴定
冷冻电子显微镜(cryo-EM)方法的改进使其在生物学和药物设计的配体结合结构测定中非常有用。然而,确定配体的构象和身份是具有挑战性的分辨率典型的低温电镜。自动化方法可以帮助配体构象建模,但目前的配体识别工具-为x射线晶体学数据开发-在低温电镜常见的分辨率下表现不佳。在这里,我们提出了EMERALD-ID,一种能够对接和评估小分子构象的方法,用于配体识别。EMERALD-ID准确识别了44%的常见配体,并在66%的情况下识别出密切相关的配体。然后,我们使用这个工具来发现可能的配体识别错误,以及以前未识别的配体。此外,我们展示了EMERALD-ID从各种小分子类型的定制配体文库中识别配体,包括人类代谢物和药物片段。我们的方法为低温电镜建模工具提供了有价值的补充,以提高小分子模型的准确性和质量。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Structure
生物-生化与分子生物学
CiteScore
8.90
自引率
1.80%
发文量
155
审稿时长
3-8 weeks
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


